
Abstract
Previous reports have shown that reactions occurring in the microdroplets formed during electrospray ionization can, under the right conditions, exhibit significantly greater rates than the corresponding bulk solution-phase reactions. The observed acceleration under electrospray ionization could result from a solution-phase, a gas-phase, or an interfacial reaction. This study shows that a gas-phase ion/molecule (or ion/ion) reaction is not responsible for the observed rate enhancement in the particular case of the Fischer indole synthesis. The results show that the accelerated reaction proceeds in the microdroplets, and evidence is provided that an interfacial process is involved.

<!-- [INSERT GRAPHICAL ABSTRACT TEXT HERE] -->
Similar content being viewed by others


Introduction
In the course of spray-based ionization, such as desorption electrospray ionization (DESI) [1] or paper spray ionization [2], derivatization reactions can be performed in order to improve the limits of detection for compounds that are difficult to ionize [3–5]. This is done during DESI by adding a reagent to the primary spray solvent, which desorbs the analyte from the surface and into the secondary droplets, where the reaction occurs during passage to the mass spectrometer (MS) [6]. In paper spray ionization, reactions are often performed by adding reagents to the paper substrate prior to analysis, and the reaction either occurs in the thin film on the paper or in the droplets after they are emitted from the paper tip [7–10]. Derivatizing reagents for ambient ionization mass spectrometry, organized by specific functional group, have been cataloged in a review article [3].
Electrospray ionization mass spectrometry (ESI-MS) is gaining recognition as a robust tool for reaction monitoring [11–15] and, alternatively, as a method for rapid screening of reactions to establish whether products are generated [16, 17]. In the first application, reaction products are simply sampled from the reaction mixture, whereas in the latter, products can be generated during the ESI event. Different conditions (initial droplet size, solvent, concentrations of reagents, droplet flight time, etc.) control these two particular outcomes. Numerous reactions (a score or so) have been found to have significantly faster rates in ESI droplets when compared with the corresponding bulk-phase reactions, and a number of these reactions have been cataloged in recent review articles [12, 18]. Amongst these accelerated reactions are quinolone and isoquinolone synthesis [19], Hantzsch synthesis of 1,4-dihydropyridines [20], hydrazone formation [6, 21], and Claisen-Schmidt condensation [22, 23]. The studies of the effects of pH [24], concentration [7] and surface activity [25] (a description of the likelihood of a given species being at the surface of a droplet) in small droplet systems suggested that the underlying cause of reaction rate acceleration lies in surface effects. Studies using acoustically levitated droplets [26], Leidenfrost levitated droplets [27], microfluidics [28], thin films [29], and “on water” chemistry [30, 31] have helped characterize acceleration phenomena in confined volumes.
It is known that the pH at the surface of electrosprayed droplets decreases [32–34] during the electrospray ionization (ESI) process. Droplets likely undergo desolvation and fission during their flight time [35–38]. Correlations between surface activity and reaction acceleration in ESI support the hypothesis that reaction acceleration is connected with surface activity [39, 40]. The main models of ESI for small molecules (the charge residue and ion evaporation models [36, 39–41]) suggest that at some point in the process of ionization, surface active molecules exist in a partially solvated form. In the charge residue model, solvent molecules continuously evaporate to yield dry ions, so there is presumably a point in the process when ions are partially solvated. In the alternative ion evaporation model, ions exist at the surface of charged droplets, where they are thought to be partially solvated at the air–droplet interface; dry ions are subsequently ejected by Coulombic forces [40]. In some cases, it has been shown that the acceleration of reactions in electrospray droplets requires that the distance between the nESI emitter and the ion transfer capillary be increased well beyond normal operating distances [20, 21, 24]; therefore, observed acceleration factors in chemical reactions under these conditions are thought to be due to reactions of partially solvated ions (increased distance between the nESI emitter and ion transfer capillary corresponds to more time for droplet evaporation as well as time for reaction). However, a gas-phase reaction mechanism is also possible since gas-phase ion/molecule reactions are much faster than solution phase reactions [42].
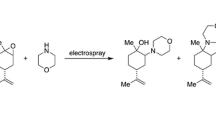
This study explores the possible contributions of a gas-phase mechanism by examining a reaction that yields different products in the gas phase from the solution phase. The Fischer indole synthesis was selected for its well documented and distinctive gas-phase ion chemistry [43, 44]. In the gas phase, phenylhydrazine (1) and acetone (2), when combined, immediately form the protonated acetone phenylhydrazone (3a). This species has been reported to instantly cyclize to (3b) based on evidence from mass spectrometry [43, 44]. This cyclization leaves the isomeric species 3a and 3b indistinguishable. Evidence for this proposal comes from gas-phase collision-induced dissociation (CID) of the ion 3b, which gives a fragment ion shown to have the same structure as protonated methylindole (4). Interestingly, under the conditions used here, the solution-phase reaction does not proceed to 4, even upon overnight reflux; instead, it forms a mixture of isomeric enamine and imine products (5 & 6) (Scheme 1). The solution-phase formation of 5 and 6 is presumably due to excess acetone in solution.

Two routes by which phenylhydrazine (1) and acetone (2) react in two phases
Experimental
An LTQ and LTQ-XL-Orbitrap (Thermo Scientific, San Jose, CA, USA) fitted with nanoelectrospray ionization (nESI) [45] sources were used for these experiments. The nESI emitter was a borosilicate glass capillary (1.5 mm o.d., 0.86 mm i.d.; Sutter Instruments, Novato, CA, USA) pulled to a tip of approximately 5 μm. A potential of 2.0 kV was applied to the inserted platinum wire to produce an electrospray. Experiments in which the emitter to MS entrance distance was varied (Figure 1) were performed using a modified moving stage with a 3D printed electrode holder to maintain alignment (See Figure S1 in Supplementary Information).

Effect of distance between the sprayer and ion transfer capillary on the progress of the Fischer indole synthesis reaction. At greater distance, the products are more abundant than at shorter distances, indicating that the electrospray process is accelerating the reaction
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified. Reactions were prepared by combining 10 μL phenylhydrazine (Eastman, Kingsport, TN, USA), 200 μL acetone, and 50 μL 1.0 M HCl in methanol (prepared by diluting 37% HCl (Mallinckrodt, St. Louis, MO, USA) in methanol to 1.0 M). An excess of acetone was used unlike a standard Fischer indole synthesis. The excess acetone allows the second (and, upon additional reflux of 48 h, the third) acetone addition(s), which compete with and prevent the formation of the standard methyl indole product (4).
Results and Discussion
Collision-Induced Dissociation of 3b
Analysis by nESI of the freshly prepared reaction mixture yielded a signal corresponding to 3b at m/z 149, which dominates the full-scan mass spectrum (Figure 2a). This reaction product was isolated and fragmented using CID, and it lost ammonia to produce the previously reported [43, 44] methyl indole product ion, 4 (Figure 2b). The ion at m/z 134 is thought to be an impurity arising from the reaction of residual aniline in the stock phenylhydrazine with acetone to form the corresponding imine. It is also possible—and more likely—that this signal arises from in situ formation of aniline from phenylhydrazine followed by aniline’s subsequent reaction with acetone. The molecular formula of 4 was verified using an LTQ-XL-Orbitrap, which confirmed the assignment C9H12N+ with an error of 0.119 ppm. The MS/MS scan provided a spectrum identical to authentic material prepared from aniline and acetone (Supplementary Figure S2).

(a) Full scan mass spectrum of freshly prepared reaction mixture with base peak corresponding to 3 and with 5 and/or 6 as well as m/z 134 resulting from a side reaction between acetone and aniline. (b) Product ion scan of isolated m/z 149 with a 1.0 Thomson isolation width and a normalized collision energy of 25 [arb units] produces primarily compound 4
The role of a possible gas-phase reaction in the open air (prior to entering the ion transfer capillary) was then explored. This was done by allowing acetone and phenylhydrazine vapor to mix from two separate, open vials. A corona discharge was struck between a platinum electrode and the ion transfer capillary to facilitate atmospheric pressure chemical ionization (APCI). The full scan mass spectrum showed 3 as the dominant species, indicating that the gas-phase reaction produces only this product. The absence of ions 5 and 6 even in the more selective MS/MS mode is noted. A product ion scan from isolated 3b produces methylindole (4) by elimination of ammonia as expected.
Solution Phase Imine/Enamine Formation
Reactions performed in the bulk solution phase were analyzed after 2 d of standing at room temperature (Figure 3a). Analysis of the solution by MS showed that the reaction provides 5 and/or 6. This demonstrates that the reaction in solution does not proceed to methylindole over time but instead favors enamine formation (5 and/or 6), represented in the mass spectrum as m/z 189.

(a) Full scan MS of the reaction mixture allowed to stand for 48 h provides the ions 5 and/or 6 exclusively. (b) Product ion scan of isolated m/z 189 shows the loss of ammonia (7) and the loss of 56 (8) as explained in Scheme 2
When the reaction mixture was heated to reflux (24 h), it yielded mainly the ion at m/z 189, as well as the product of an additional acetone addition at m/z 229 (see Supplemental Information). Further reflux (48 h) and addition of acetone promotes the conversion of m/z 189 to 229. The product ion scan of the isolated ion at m/z 189 yielded fragment ions due to the loss of neutral species with masses of 17 and 56 Da. The loss of 17 corresponds to the loss of ammonia, yielding the acetone imine of methylindole (8). The loss of 56 is not a subsequent fragmentation of the enamine of methylindole (confirmed by MS3); rather, it corresponds to protonated 2-aminoindole (7). The suggested structure of compound 7 arises from a postulated fragmentation mechanism, whereby a rearrangement is followed by elimination of butene as shown in Scheme 2.

Postulated fragmentation mechanism to form 2-amino indole (7) by rearrangement followed by loss of neutral butene. The charge site is not known
Compound 7 has previously been reported as a product of thermal activation of an intermediate species in the reaction of phenylhydrazine and acetone [44] when nebulized by electrospray; however, the phase in which it forms (solution/gas phase) had not been delineated. This work shows that a solution-phase mechanism was likely to be responsible for the formation of the precursor ion that fragments to produce 7 in the previous literature.
Reaction Acceleration in Charged Droplets
A significant excess of acetone was required to observe accelerated formation of the methyl amino-indole (3) with distance. This is most likely due to the volatility of acetone compared with the phenylhydrazine and methanol. At lower concentrations, the acceleration with distance decreases and eventually ceases. This suggests that, as expected, nESI can be used safely to monitor reaction kinetics provided the limitations imposed by possible reaction acceleration are recognized and avoided. The acceleration effect can be seen in Figure 4, and the acceleration factor is 10 if the m/z 189 (corresponding to 5/6) is added back. That is, the acceleration factors was estimated as the ratio of product (5 + 6 + 7)/starting material (3) in the droplet phase as opposed to bulk solution. This acceleration shows that these accelerated reactions favor a solution-phase droplet mechanism over an alternative gas-phase mechanism. This is evident from the fact that the two reagents in the gas phase produced 3 and not 5/6; the ESI droplet experiment (using extended distance between spray emitter and ion transfer capillary) produced accelerated formation of 5 and/or 6 with respect to 3.

NanoESI of fresh reaction mixture at a distance of 0.5 cm (a) and 7 cm (b) between the ion transfer capillary and the tip of the nESI source shows an increase in the abundance of 5 and/or 6 with respect to 3 (an impurity reacting with acetone at m/z 134 and the fragment of 5 (7) are also present in the full scan spectrum). The spectra are each an average of 10 scans at unit resolution
In an effort to probe reaction at the surface of the droplet, two different surfactants (triton x-100 and pentadecanoic acid) were doped into the reaction mixture. Fresh reaction mixtures were prepared and surfactant was added to the mixture at 1% v/v before analysis. The spectra acquired in the presence of surfactant were of lower quality and contained many interfering peaks; however, the peaks of interest were significant and reproducible in the MS full scan. Surfactants reduced the surface tension of the droplet, creating smaller droplets sooner [46]. This is because the Rayleigh limit (the point at which droplets undergo fission) is reached faster as the surface tension is reduced. Surfactants decrease surface tension and the distance required to achieve equal acceleration effects without surfactants falls from 7 to 3 cm. Interestingly, as the distance was increased further (to the 7 cm distance, which yielded acceleration without surfactant, the spectrum recorded using surfactant was again dominated by 3. This is interpreted as indicating a contribution of a gas-phase reaction to the observed mass spectrum, as the droplets were allowed to completely desolvate, yielding dry reagent ions, which produced the gas phase product, 6. This result was confirmed to be an artifact of the spray process and not due to bulk-phase reaction by returning the sprayer to the 3 mm distance and repeating the distance experiment and acquiring the same result as previously discussed. This observation is significant as it bolsters the hypothesis that the observed acceleration does indeed proceed via a solution-phase mechanism.
Conclusion
The formation of reaction products 5 and 6 can be accelerated by increasing the distance between the nESI emitter and the ion transfer capillary. As this variant of the Fischer indole synthesis forms a different final product in solution as opposed to gas phase, it has been shown that the reaction accelerated in ESI favors the solution-phase product over the ion-molecule gas-phase products. When surfactants are added, acceleration can be achieved at a smaller distance between the ion transfer capillary and the nESI emitter compared with the reactions accelerated without surfactants, pointing to the importance of surface reactions.
References
Takáts, Z., Wiseman, J.M., Gologan, B., Cooks, R.G.: Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306(5695), 471–473 (2004)
Liu, J., Wang, H., Manicke, N.E., Lin, J.-M., Cooks, R.G., Ouyang, Z.: Development, characterization, and application of paper spray ionization. Anal. Chem. 82(6), 2463–2471 (2010)
Espy, R.D., Wleklinski, M., Yan, X., Cooks, R.G.: Beyond the flask: reactions on the fly in ambient mass spectrometry. TrAC Trends Anal. Chem. 57, 135–146 (2014)
Wu, C., Ifa, D.R., Manicke, N.E., Cooks, R.G.: Rapid, direct analysis of chloesterol by charge labeling in reactive desorption electrospray ionization. Anal. Chem. 81, 7618–7624 (2009)
Wu, Q., Comi, T.J., Li, B., Rubakhin, S.S., Sweedler, J.V.: On-tissue derivatization via electrospray deposition for matrix-assisted laser desorption/ionization mass spectrometry imaging of endogenous fatty acids in rat brain tissues. Anal. Chem. 88(11), 5988–5995 (2016)
Girod, M., Moyano, E., Campbell, D.I., Cooks, R.G.: Accelerated bimolecular reactions in microdroplets studied by desorption electrospray ionization mass spectrometry. Chem. Sci. 2(3), 501–510 (2011)
Yan, X., Augusti, R., Li, X., Cooks, R.G.: Chemical reactivity assessment using reactive paper spray ionization mass spectrometry: the Katritzky reaction. ChemPlusChem 78(9), 1142–1148 (2013)
Zhou, X., Pei, J., Huang, G.: Reactive paper spray mass spectrometry for in situ identification of quinones. Rapid Commun. Mass Spectrom. 29(1), 100–106 (2015)
Bag, S., Hendricks, P., Reynolds, J.C., Cooks, R.G.: Biogenic aldehyde determination by reactive paper spray ionization mass spectrometry. Anal. Chim. Acta 860, 37–42 (2015)
Mazzotti, F., Di Donna, L., Taverna, D., Nardi, M., Aiello, D., Napoli, A., Sindona, G.: Evaluation of dialdehydic anti-inflammatory active principles in extra-virgin olive oil by reactive paper spray mass spectrometry. Int. J. Mass Spectrom. 352, 87–91 (2013)
Vikse, K.L., Woods, M.P., McIndoe, J.S.: Pressurized sample infusion for the continuous analysis of air- and moisture-sensitive reactions using electrospray ionization mass spectrometry. Organometallics 29(23), 6615–6618 (2010)
Ingram, A.J., Boeser, C.L., Zare, R.N.: Going beyond electrospray: mass spectrometric studies of chemical reactions in and on liquids. Chem. Sci. 7(1), 39–55 (2016)
Ma, X., Zhang, S., Zhang, X.: An instrumentation perspective on reaction monitoring by ambient mass spectrometry. TrAC Trends Anal. Chem. 35, 50–66 (2012)
Yan, X., Bain, R.M., Li, Y., Qiu, R., Flick, T.G., Cooks, R.G.: Online inductive electrospray ionization mass spectrometry as a process analytical technology tool to monitor the synthetic route to anagliptin. Org. Process Res. Dev. 20(5), 940–947
Yan, X., Sokol, E., Li, X., Li, G., Xu, S., Cooks, R.G.: On-line reaction monitoring and mechanistic studies by mass spectrometry: Negishi cross-coupling, hydrogenolysis, and reductive amination. Angew. Chem. Int. Ed. Engl. 53(23), 5931–5935 (2014)
Santanilla, A.B., Regalado, E.L., Pereira, T., Shelvin, M., Bateman, K., Campeau, L., Schneeweis, J., Berritt, S., Shi, Z., Nantermet, P., Liu, Y., Helmy, R., Welch, C.J., Vachal, P., Davies, I.W., Cernak, T., Dreher, S.D.: Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science 347(6217), 49–53 (2015)
Wleklinski, M., Falcone, C.E., Loren, B.P., Jaman, Z., Iyer, K., Ewan, H.S., Hyun, S.-H., Thompson, D.H., Cooks, R.G.: Can accelerated reactions in droplets guide chemistry at scale? European J. Org. Chem. 2016(33), 5480–5484 (2016)
Yan, X., Bain, R.M., Cooks, R.G.: Organic Reactions in Microdroplets: Reaction Acceleration Revealed by Mass Spectrometry. Angew. Chem. Int. Ed. Engl. (2016)
Lee, J.K., Banerjee, S., Nam, H.G., Zare, R.N.: Acceleration of reaction in charged microdroplets. Q. Rev. Biophys. 48(4), 437–444 (2015)
Bain, R.M., Pulliam, C.J., Cooks, R.G.: Accelerated Hantzsch electrospray synthesis with temporal control of reaction intermediates. Chem. Sci. 6(1), 397–401 (2015)
Bain, R.M., Pulliam, C.J., Ayrton, S.T., Bain, K., Cooks, R.G.: Accelerated hydrazone formation in charged microdroplets. Rapid Commun. Mass Spectrom. 30(16), 1875–1878 (2016)
Muller, T., Badu-Tawiah, A., Cooks, R.G.: Accelerated carbon–carbon bond-forming reactions in preparative electrospray. Angew. Chem. Int. Ed. Engl. 51(47), 11832–11835 (2012)
Bain, R.M., Pulliam, C.J., Yan, X., Moore, K.F., Müller, T., Cooks, R.G.: Mass spectrometry in organic synthesis: Claisen-Schmidt base-catalyzed condensation and Hammett correlation of substituent effects. J. Chem. Edu. 91(11), 1985–1989 (2014)
Lee, J.K., Kim, S., Nam, H.G., Zare, R.N.: Microdroplet fusion mass spectrometry for rast reaction kinetics. Proc. Natl. Acad. Sci. U. S. A. 112(13), 3898–3903 (2015)
Li, Y., Yan, X., Cooks, R.G.: The role of the interface in thin film and droplet accelerated reactions studied by competetive substituent effects. Angew. Chem. Int. Ed. 55, 3433–3437 (2016)
Crawford, E.A., Esen, C., Volmer, D.A.: Real time monitoring of containerless microreactions in acoustically levitated droplets via ambient ionization mass spectrometry. Anal. Chem. 88(17), 8396–403 (2016)
Bain, R.M., Pulliam, C.J., Thery, F., Cooks, R.G.: Accelerated chemical reactions and organic synthesis in Leidenfrost droplets. Ang. Chem. Int. Ed. Engl 55(35), 10478–10482 (2016)
Fallah-Araghi, A., Meguellati, K., Baret, J.C., El Harrak, A., Mangeat, T., Karplus, M., Ladame, S., Marques, C.M., Griffiths, A.D.: Enhanced chemical synthesis at soft interfaces: a universal reaction-adsorption mechanism in microcompartments. Phys. Rev. Lett. 112(2), 028301 (2014)
Badu-Tawiah, A.K., Campbell, D.I., Cooks, R.G.: Accelerated C–N bond formation in dropcast thin films on ambient surfaces. J. Am. Soc. Mass Spectrom. 23(9), 1461–1468 (2012)
Narayan, S., Muldoon, J., Finn, M.G., Fokin, V.V., Kolb, H.C., Sharpless, K.B.: “On water”: unique reactivity of organic compounds in aqueous suspension. Angew. Chem. Int. Ed. Engl. 44(21), 3275–3279 (2005)
Jung, Y., Marcus, R.A.: On the theory of organic catalysis “on Water”. J. Am. Chem. Soc. 129(17), 5492–5502 (2007)
Wang, G., Cole, R.B.: Effect of solution uonic strecgth on analyte charge state distributions in positive and negative ion electrospray mass spectrometry. Anal. Chem. 33, 3702–3708 (1994)
Zhou, S., Prebyl, B.S., Cook, K.D.: Profiling pH changes in the electrospray plume. Anal. Chem. 74, 4485–4888 (2002)
Fenn, J.B.: Ion formation from charged droplets: roles of geometry, energy, and time. J. Am. Soc. Mass Spectrom. 4, 524–535 (1993)
Wortmann, A., Kistler-Momotova, A., Zenobi, R., Heine, M.C., Wilhelm, O., Pratsinis, S.E.: Shrinking droplets in electrospray ionization and their influence on chemical equilibria. J. Am. Soc. Mass Spectrom. 18(3), 385–393 (2007)
Tang, K., Gomez, A.: On the structure of an electrostatic spray of monodisperse droplets. Phys. Fluids 6(7), 2317 (1994)
Wilhelm, O., Mädler, L., Pratsinis, S.E.: Electrospray evaporation and deposition. J. Aerosol Sci. 34(7), 815–836 (2003)
Smith, J.N., Flagam, R.C., Beauchamp, J.L.: Droplet evaporation and discharge dynamics in electrospray ionization. J. Phys. Chem. A 106, 9957–9967 (2002)
Cech, N.B., Enke, C.G.: Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 20(6), 362–387 (2001)
Konermann, L., Ahadi, E., Rodriguez, A.D., Vahidi, S.: Unraveling the mechanism of electrospray ionization. Anal. Chem. 85(1), 2–9 (2013)
Yue, X., Vahidi, S., Konermann, L.: Insights into the mechanism of protein electrospray ionization from salt adduction measurements. J. Am. Soc. Mass Spectrom. 25(8), 1322–1331 (2014)
Gioumousis, G., Stevenson, D.P.: Reactions of gaseous molecule ions with gaseous molecules. V. Theory. J. Chem. Phy. 29(2), 294–299 (1958)
Glish, G.L., Cooks, R.G.: The Fischer indole synthesis and pinacol rearrangement in the mass spectrometer. J. Am. Chem. Soc. 100(12), 6720–6725 (1978)
Chen, H., Eberlin, L.S., Nefliu, M., Augusti, R., Cooks, R.G.: Organic reactions of ionic intermediates promoted by atmospheric-pressure thermal activation. Angew. Chem. Int. Ed. Engl. 47(18), 3422–3425 (2008)
Wilm, M., Mann, M.: Analytical properties of the nanoelectrospray ion source. Anal. Chem. 68(1), 1–8 (1996)
Badu-Tawiah, A., Cooks, R.G.: Enhanced ion signals in desorption electrospray ionization using surfactant spray solutions. J. Am. Soc. Mass Spectrom. 21(8), 1423–1431 (2010)
Acknowledgements
The authors acknowledge the financial assistance of the National Science Foundation (CHE-1307264).
Author information
Authors and Affiliations
Corresponding author
Additional information
Ryan M. Bain and Stephen T. Ayrton contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 479 kb)
Rights and permissions
About this article

Cite this article
Bain, R.M., Ayrton, S.T. & Cooks, R.G. Fischer Indole Synthesis in the Gas Phase, the Solution Phase, and at the Electrospray Droplet Interface. J. Am. Soc. Mass Spectrom. 28, 1359–1364 (2017). https://doi.org/10.1007/s13361-017-1597-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1597-z