
Abstract
We present the first application of the quality threshold (QT) clustering algorithm to mass spectrometry (MS) data. The unique abilities of QT clustering to yield precision nodes that are commensurate with the mass measurement precision of the instrument are exploited to generate a consensus spectrum out of multiple replicate spectra. The spectral dot product and confidence intervals are used as a tool for evaluating the similarity and reproducibility between the consensus and replicates. The method is equally applicable to high and low resolution measurements. This paper demonstrates applications to linear spectra from a matrix assisted laser desorption ionization (MALDI) time of flight (TOF) instrument as well as peptide fragmentation data obtained from a TOF/TOF after unimolecular decomposition. The advantages of clustering to mitigate the inherent precision the shortcomings of MALDI data are discussed.
Similar content being viewed by others

1 Introduction
Since the development in the 1980s by Karas and Hillenkamp [1] of a laser desorption ionization employing small organic molecules as UV absorbers, MALDI has an ionization method that is applied successfully to analysis of a huge range of molecules, including intact proteins [1–3] and synthetic polymers [4] to peptides in general [5, 6] as well as part of studies in proteomics [7–10]. In addition, the method has been applied to measurements of polynucleotides [11–14] as well as a large number of smaller molecules, including lipids [15–17], fatty acids, bile acids, and steroids [18–20].
The laser desorption ionization basis of MALDI gives rise to ion formation processes that are exceedingly complex and, despite more than 20 years of investigation and modeling, are not yet completely understood [21–23]. Complicating any fundamental aspects of ion formation are the well known and rather subjective issues of the “right” matrix for any analyte [1, 11, 13, 15, 18, 24–28], including the use of ionic liquid matrices [29, 30]. In addition, there have been efforts directed towards the “proper” method for sample preparation [8, 21]. The various approaches to matrix use and sample preparation have all been more or less effective for particular applications, especially when they lead to greater homogeneity of the analyte/matrix mixtures.
While not denying the importance of these efforts to understand ionization fundamentals or the effects of different matrices and sample preparation on spectral quality, the ultimate goal of all these studies has been the generation of analytically useful mass spectra. However, for any given sample prepared in a given manner, it is well known that there is extensive variability among multiple replicate spectra. Thus, a method to address this experimental variance in a consistent fashion in order to produce truly reliable spectra becomes necessary. This problem becomes particularly important in the analysis of complex samples because such spectra contain peaks that are inconsistent in both their occurrence and intensity. It is true that obtaining multiple spectra for each sample is valuable to assess the variance in the measurements, but evaluating differences between the replicates manually can be difficult if not impossible. In this work, we present an improved method for generating reliable MALDI spectra through the acquisition of multiple replicates and the formation of a consensus spectrum. Earlier efforts to evaluate mass spectral reproducibility have either relied on binning mass-to-charge intensities [31, 32] or k-means clustering with an a priori determined number of nodes [33]. As high resolutions are possible in MALDI spectra, mass binning is inappropriate except for specific cases in which the peaks have a uniform mass defect, the mass precision is small compared with the size of the bin, and only one peak is expected for each bin. As MALDI spectra have a variable number of peaks, the predetermination of a number of nodes to perform k-means clustering is inappropriate. The use of k-means clustering is also problematic because there is no quality guarantee on the results, so the mass accuracy of the clustered peak list can be significantly lower than the mass accuracy of the replicate spectra that are used to derive it. As an alternative, the approach presented here employs QT clustering [34] of similar m/z values within a reasonable mass precision without any other assumption on the data. Other investigators have used hierarchical clustering to select which spectra in very large data sets can be “merged” or clustered [35]. In contrast, this work uses replicate spectra and forms clusters of peaks within those spectra for subsequent analysis. The dot product-based statistical evaluations of consensus and replicate validated in our earlier work continue to be used, but instead of generating intensity vectors based on mass bins, the vectors are based on precision nodes. The dot product is used as a similarity metric to evaluate the suitability of including a replicate in the set of replicates that generate the consensus spectrum. As this method takes into account the precision associated with every peak in the spectrum, it is equally applicable to reflector and linear TOF spectra, and we also demonstrate its use to improve the results from de novo sequencing of TOF/TOF peptide fragmentation spectra.
2 Methods
2.1 Reagents
Deionized water (Hydroservices, Durham, NC, USA), acetonitrile (ACN; Burdick and Jackson, HPLC Grade; Honeywell B and D, Muskegon, MI, USA), trifluoroacetic acid (TFA; Fluka Chemicals, Milwaukee, WI, USA) were used as received; a-cyano hydroxycinammic acid (CHCA; Sigma-Aldrich, Milwaukee, WI, USA) was recrystallized from ethanol and frozen aliquots reconstituted and used as required; a 10 M cyanogen bromide (CNBr, Sigma-Aldrich, Milwaukee, WI, USA) solution in ACN was prepared as stock and stored at 4 °C in a light tight tube for use in digestions. ACTH peptides (Sigma-Aldrich) used as internal standards were prepared as 1 uM solutions in 1:1 ACN:0.1% TFA and frozen for later addition to matrix solutions; synthetic tubulin peptide was synthesized for D. Sackett and RfffR peptide (f = pentafluoro-phenylalanine) was synthesized by the Johns Hopkins Peptide Synthesis Facility, and both were prepared and used similarly to the ACTH peptides.
2.2 Instrument
An AB SCIEX model 4800 TOF/TOF (AB SCIEX, Foster City, CA, USA) was used for all measurements using positive ion MS/MS mode, collision gas off, or negative ion linear mode. Data were acquired under instrument control and results transferred to a remote computer for subsequent processing using the T2dExtractor tool that interacts with the Oracle database on the 4800 [36].
2.3 Peptide Fragmentation
Peptides from a solution tryptic digestion of BSA were prepared in a 10 nM solution of 1:1 ACN:0.1% TFA and were spotted as mixtures in 0.5 uL (500 fmol) aliquots along with an equal volume of matrix (CHCA, 5 mg/mL in 50% ACN with 10 mM NH4H2PO3 to suppress matrix cluster ions) containing internal standards, RfffR ([MH]+, 1042.276 ) and ACTH 18–39 clip ([MH+, 2465.199) from each of three sample wells. Internally calibrated positive ion reflector spectra were acquired and were used to select five peptides for fragmentation: m/z 927.494, 1439.812, 1479.796, 1567.743, and 1639.938. Positive ion MS/MS spectra were acquired in gas off mode using a default calibration established using five y-series ions from the fragmentation of Glu1-fibrinopeptide B and requiring three successive default calibration updates with less than 1% change in calibration parameters and maximum outlier calibration values of less than 0.025 Da. Six replicate 1000 shot MS/MS spectra for each peptide were acquired from each sample well with no apparent diminution of fragment ion intensities; these six were used for the analysis described below.
2.4 Tubulins
Tubulin proteins were extracted from rat brains (RBT) by an established method [37–39] and cleaved relative to methionine using an optimized protocol involving 0.1 M CNBr, roughly a 10,000-fold molar excess, in 20% TFA solution. Cleavage proceeded overnight at room temperature in a light tight vessel. After cleavage, the sample was quenched with an equal volume of deionized water, dried using a Speed Vac (Savant; Thermo-Fisher Scientific), Pittsburgh, PA redissolved in water, and redried; peptides were redissolved and the C-terminal peptides separated from peptides from the globular region of the protein using strong anion exchange (SAX). The SAX eluate was dried and redissolved in 10 uL 1% TFA and spotted into two sample wells; matrix 5 mg/mL CHCA in 50% ACN with 10 mM (NH4)2SO4 to suppress sodium ion adduction [40]); the matrix solution also contained mass standards for internal calibration as average m/z values: synthetic human a-tubulin ([M – H]–, m/z 1962.91), ACTH 18–39 clip ([M – H]–, m/z 2464.66), ACTH 1–39 ([M – H]–, m/z 4541.07). In order to obtain reliable spectral intensities for minor components of the C-terminal peptide mixtures, samples were spotted by adding 1 uL sample, 0.5 uL matrix and then as samples dried two additional 0.5 uL aliquots of sample were added to each well. Nine replicate 1000 shot spectra were acquired in linear negative ion mode, mass range 1500–6000 Da, from each well.
2.5 Calculations
2.5.1 Clustering
Data were migrated as peak lists of m/z and S/N from the mass spectrometer Oracle database to a desktop computer for further analysis using the T2d Extractor tool. While the term spectrum is used extensively throughout this paper, it should be noted that rather than full spectra, peak lists are used as a surrogate. The S/N values were normalized to a unit vector and read into an R-script containing the QT clustering algorithm [34] in the flexclust package that is available from cran.R-project.org. Prior to this work, the algorithm has been used only for determining clusters of genes within genetic databases. The algorithm starts by making each peak into a candidate node. All the other peaks with a difference in mass less than the instrumental precision are clustered into the candidate node. This results in a highly redundant set of nodes that is simplified by selecting the non-redundant set in that incorporates the largest nodes. Since this algorithm was developed and implemented for gene expression array data, it does not weight by a second dimension, intensity weighting was achieved by listing the masses in the array redundantly (the mass was listed a number of times equal to the integer value of its intensity divided by the minimum intensity). The algorithm requires the specification of a “quality threshold,” which in the context of gene expression data refers to the largest acceptable value for the distance metric, e.g., Euclidian, Manhattan, etc., between two genes in the same candidate node. In the context of this work the quality threshold refers to the mass measurement precision. The mass measurement precision was validated previously to be 0.05 Da for TOF/TOF spectra and 1.0 Da for linear TOF measurements. A consensus peak list was generated by selecting only the clusters that were present in at least half of the peak lists. The peak masses of the replicate spectra were mapped to the consensus spectrum to enable the calculation of the replicate-versus-consensus dot products [31]. This process is summarized in Scheme 1.

Flowchart of computation steps for production and use of consensus spectra
2.5.2 Analysis of Peptide Fragmentation Spectra
Individual peptide fragmentation spectra along with the consensus spectra developed from them were subjected to de novo sequencing using the MAARIAN algorithm described previously [41]. For this work, peak lists were migrated from the 4800 Oracle database as Mascot generic files that were modified by addition of mass and intensity data for the precursor ion since this is ion is not considered in Mascot searches. Input parameters for MAARIAN were set to allow intensities of all peaks in any given list to be used allowing one missed fragmentation except for the m/z 1479 ion for which two missed fragmentations were required. All sequences were searched with a tolerance of 0.05 Da. There are no other parameters to adjust in MAARIAN.
2.5.3 Analysis of Tubulin Spectra
Individual RBT spectra along with the consensus spectrum were analyzed for the best match between them and a list of average masses for all possible RBT species using a Perl script written for the purpose and described in a previous publication [31]. The script produces a list of observed masses that are matched, the mass error of the match, and the relative intensity of the peak matched. The list used for matching was generated from the rat tubulin genes shown in Table 1 and consists of a compilation of CNBr cleavage products listed as [M – H]– ions at average mass values including up to one missed cleavage. The list also contains predicted C-terminal modifications: up to 12 glutamylations, and detyrosination of alpha subunits with and without the removal of the penultimate glutamate. This leads to a list of 879 candidate masses for matching against the consensus spectrum. A 1.0 Da tolerance was used for all matches.
3 Results and Discussion
3.1 Peptide Fragmentation
Replicate fragmentation spectra for each of the five peptides in each of the three wells were processed by the clustering algorithm. Each replicate and the consensus spectra were then sequenced de novo. Results for one well are summarized in Table 2, and results for the other two wells were virtually identical. With the exception of the YLYEIAR and KVPQVSTPTLVEVSR peptides, Table 2 shows that having MAARIAN determine the correct sequence is the exception rather than the rule, and even for those two peptides, any single replicate cannot be expected to necessarily yield a correct result. Despite this rather substantial variation, formation of the consensus spectra gave a correct sequence in every instance aside from a low level of sequence ambiguity that can be readily accounted for in subsequent analysis of a protein. It is interesting to note that use of our earlier clustering algorithm that used mass-binning to form the consensus spectra gave results that were slightly less successful than the QT clustering approach (data not shown).
It should be noted that fragmenting peptides by unimolecular decomposition leads to spectra that have subtle, mainly qualitative, differences. Thus, the fragmentation spectra discussed here are commensurate with other fragmentation spectra and have uniformly high (>0.85) dot products with no non-correlating spectra. This effect is the same regardless of whether the precursor is included or excluded in the normalized spectrum (data not shown). As a result, the benefit of QT clustering does not derive as much from quantitative effects such as the refinement of mass and intensity precision as from the qualitative inclusion or exclusion of low intensity but reproducible peaks that clarify ambiguous sequence results. While none of the replicates are rejected on the basis of a negative dot product, the exclusion of irreproducible peaks from the final consensus spectrum yields a final product that gives full sequence information. For example, in the extreme case of the RHPEYAVSVLLR peptide, more than half of the spectra have useful sequencing peaks, but no individual replicate contains the full sequence. Incorporation of all of the robust features into a single spectrum is necessary to obtain the full correct sequence.
3.1.1 Tubulins
The results of performing the clustering and reproducibility evaluations on 30 replicate tubulin spectra acquired in groups of 10 from three sample wells are shown in Table 3. The first column shows the average percentage of total intensity assigned for raw spectra, and the second column shows this assignment as a result of aligning each spectrum to the masses found in the consensus. As QT clustering is an alternative method of spectral alignment, the results shown in the second column show only modest increases to total intensity assigned simply by mapping the individual replicate spectra to those masses. However, since mass variance is a relatively small portion of overall MALDI variance—even in this relatively low precision example of linear spectra—the improvement seen from assigning the consensus spectrum, column 3 of Table 3, is appreciably greater than the improvement from simply aligning masses. In addition, using the consensus spectrum to identify poor quality spectra (those spectra that have a negative lower 95% confidence interval on the spectral dot product with the consensus spectrum [31]) allows for a straightforward objective way to exclude these spectra and raise the average and range of identifiable peaks. This is shown in columns 4 and 5 of Table 3; column 4 shows the effect of removing uncorrelated spectra and considering replicates based on mass alignment while the last column shows the effect of forming the consensus spectrum with the uncorrelated replicates removed: the result is a consistently high level of reproducibility and spectral quality. It is particularly noteworthy to see the consequence of excluding the uncorrelated spectra for the Set 3 data, a set of spectra that clearly contains a number (4/9) of rather noisy replicates. Comparing the final values in column 5 of Table 3 to the range maxima in column 1 demonstrates little difference from the best spectra at the beginning of the algorithm and the consensus spectra at the end. As it is impossible to know which spectra are “good” and which are “bad,” this method provides an unsupervised way to select the best features and obtain a higher confidence in the results.
3.1.2 Overall Considerations
The computational expense of QT clustering is reported in Tables 2 and 3. While these run times are certainly not extravagant for the in-depth analysis of spectra often required for a focused workup of multiple replicates from a single biological sample, they are sufficiently large to limit the utility of this approach to small numbers (<100) of replicates. For experiments in which the number of replicates is predetermined, such as the experiments presented here, this is not a problem because acquiring more replicates takes more time, and only a small number or replicates is necessary to achieve improvement. For large, unmonitored experiments in which the number of available replicates can be orders of magnitude larger than those presented in this paper, a method of determining which representative replicates should be clustered into a consensus is prudent.
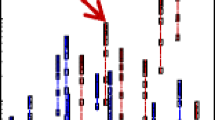
The ultimate goal of yielding reproducible data is to arrive at a sound and trusted conclusion. Thus, evaluating the quality of a data method on the basis of reproducibility metrics alone is clearly inadequate. While the assignment of peaks in a spectrum to specific functions such as peptide sequence or tubulin isotypes, a discussion of the biological meaning conferred by those assignments is a separate matter from the focus of this paper. It is important, however, to look at the impact of these algorithms on the overlap of assignments rather than the percentage of assignment alone. In the case of the tubulin spectra, Figure 1 demonstrates the differences in overlaps between individual replicates from sets 1, 2, and 3 as well as the overlaps for the consensus spectra. In Figure 1a, the three least correlating replicates from Sets 1, 2, and 3, i.e., the three lowest dot products, share only two peaks. The spectrum in Set 2 shows no other overlaps with the spectra from Sets 1 and 3 despite it having more than double the number of peaks than either of the other two. On the other extreme, Figure 1b demonstrates excellent overlap among the three replicate spectra that have the highest dot products (or correlation) with their respective consensus spectra. In this case, there are 45 peaks in common among the three replicates and many fewer peaks that are specific to only a single set. As further evidence that the consensus spectrum mitigates this broad lack of reproducibility, the consensus overlaps are shown in Figure 1c. As the reproducibility increases for the consensus spectra, the overlaps between the three groups also increase. There is both an increase in the total number of peaks and the portion of identifiable peaks in the consensus compared with the replicates of any set. Appreciably more than half of the total peaks in any one consensus are found in common in each of the consensus spectra.

A demonstration of the overlapping numbers of tubulin peptides identified by the steps of the experiments detailed in Table 3. (a) The overlap of the three least correlating discrete replicates, and designated by *, from sets 1, 2, and 3; (b) the overlap of the three best correlating discrete replicates from sets 1, 2, and 3; and (c) the overlap of consensus spectra for sets 1, 2, and 3 after pruning the uncorrelated replicates
4 Conclusion
The method presented is the first that uses QT clustering for mass spectrometry. The new desirable features of this approach include its ability to work with data that have different mass measurement precisions as well as the lack of assumptions that are made about the data entering the algorithm. Imprecision mitigation is particularly valuable for MALDI measurements because situations abound for which MALDI is particularly suited to the performance demands of the investigation. At the same time, ignoring the precision shortcomings has the potential for misleading results. In the near future, we plan to test this method on data from other mass spectrometers, including electrospray quadrupole-time of flight (ESI qTOF) hybrid instruments. A publicly available website that implements this algorithm and will allow any user to generate consensus spectra from replicates is also under development: http://MassSpectrometry.nichd.nih.gov/.
References
Karas, M., Bachman, D., Bahr, U., Hillenkamp, F.: Matrix-assisted ultraviolet laser desorption of non-volatile compounds. Int. J. Mass Spectrom. Ion Processes 78, 53–68 (1987)
Lin, M., Campbell, J.M., Mueller, D.R., Wirth, U.: Intact protein analysis by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectromety. Rapid Commun. Mass Spectrom. 17,1809–1814 (2003).
Liu, Z.Y., Schey, K.L.: Optimization of a MALDI TOF-TOF mass spectrometer for intact protein analysis. J. Am. Soc. Mass Spectrom. 16(4), 482–490 (2005)
Zhu, H., Yalcin, T., Li, L.: Analysis of the accuracy of determining average molecular weights of narrow polydispersity polymers by matrix-assosted laser desorption ionization time-of-flight mass spectrometry. J. Am. Soc. Mass Spectrom. 9, 275–281 (1998)
Chaurand, P., Luetzenkirchen, F., Spengler, B.: Peptide and protein identification by matrix-assisted laser desorption ionization (MALDI) and MALDI-post-source decay time-of-flight mass spectrometry. J. Am. Soc. Mass Spectrom. 10, 91–103 (1999)
Egelhofer, V., Büssow, K., Luebbert, C., Lehrach, H., Nordhoff, E.: Improvements in protein identification by MALDI-TOF-MS peptide mapping. Anal. Chem. 72(13), 2741–2750 (2000)
Nelson, R.W., Nedelkov, D., Tubbs, K.A.: Biosensor chip mass spectrometry: a chip-based proteomics approach. Electrophoresis 21(6), 1155–1163 (2000)
Landry, F., Lombardo, C.R., Smith, J.W.: A method for application of samples to matrix-assisted laser desorption ionization time-of-flight targets that enhances peptide detection. Anal. Biochem. 279(1), 1–8 (2000)
Berndt, P., Hobohm, U., Langen, H.: Reliable automatic protein identification from matrix-assisted laser desorption/ionization mass spectrometric peptide fingerprints. Electrophoresis 20(18), 3521–3526 (1999)
Marcus, K., Immler, D., Sternberger, J., Meyer, H.E.: Identification of platelet proteins separated by two-dimensional gel electrophoresis and analyzed by matrix assisted laser ddesorption/ioniztion-time of flight-mass spectrometry and detection of tyrosine-phosphorylated proteins. Electrophoresis 21(13), 2622–2636 (2000)
Fitzgerald, M.C., Parr, G.R., Smith, L.M.: Basic matrixes for the matrix-assisted laser desorption/ionization mass spectrometry of proteins and oligonucleotides. Anal. Chem. 65(22), 3204–3211 (1993)
Costello, C.E., Juhasz, P., Perreault, H.: New mass-spectral approaches to ganglioside structure determinations. In biological function of gangliosides—Proceedings of Nobel Symposium 83. In: Svennerholm, L., Asbury, A.K., Reisfeld, R.A., Sandhoff, K., Suzuki, K., Tettamanti, G., Toffano, G. (eds.) Progress in Brain Research, pp. 45–61. Elsevier Science Publishers B V, Amsterdam (1994)
Lecchi, P., Le, H.M.T., Pannell, L.K.: 6-Aza-A-thiothymine—a matrix for MALDI spectra of oligonucleotides. Nucleic Acids Res. 23(7), 1276–1277 (1995)
Haff, L.A., Juhasz, P., Martin, S.A., Roskey, M.T., Smirnov, I.P., Takach, E.J., Vestal, M.: DNA-sequence analysis by delayed extraxction matrix-assisted laser-desorption mass-spectrometry. Clin. Chem. 41(11), 1680–1686 (1995)
Stuebiger, G., Belgacem, O.: Analysis of lipids using 2, 4, 6-trihydroxyacetophenone as a matrix for MALDI mass spectrometry. Anal. Chem. 79(8), 3206–3213 (2007)
Jackson, S., Wang, H., Woods, A.: In situ structural characterization of glycerophospholipids and sulfatides in brain tissue using MALDI-MS/MS. J. Am. Soc. Mass Spectrom. 18(1), 17–26 (2007)
Mcdonald-Marsh, T., Carroll, C., Robinson, N., Musatov, A.: Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis of cardiolipin extracted from detergent-solubilized mitochondrial electron transfer complexes. Anal. Biochem. 359(2), 262–264 (2006)
Galesio, M., Rial-Otero, R., Capelo-Martínez, J.-L.: Comparative study of matrices for their use in the rapid screening of anabolic steroids by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 23(12), 1783–1791 (2009)
Griffiths, W.J.: Tandem mass spectrometry in the study of fatty acids, bile acids, and steroids. Mass Spectrom. Rev. 22(2), 81–152 (2003)
Griffiths, W.J., Liu, S., Alvelius, G., Sjövall, J.: Derivatization for the characterization of neutral oxosteroids by electrospray and matrix-assisted laser desorption/ionization tandem mass spectrometry: the Girard P Derivative. Rapid Commun. Mass Spectrom. 17(9), 924–935 (2003)
Dreisewerd, K.: The desorption process in MALDI. Chem. Rev. 103, 395–725 (2003)
Karas, M.; Kruger, R.: Ion formation in MALDI: the cluster ionization mechanism. Chem. Rev. 103, 427–439 (2003)
Knochenmuss, R., Zenobi, R.: MALDI ionization: the role of in-plume processes. Chem. Rev. 103, 441–452 (2003)
Jaskolla, T.W., Lehmann, W.-D., Karas, M.: 4-Chloro- ↦ -cyanocinnamic acid is an advanced, rationally designed MALDI matrix. Proc. Natl. Acad. Sci. U. S. A. 105(34), 12200–12205 (2008)
Juhasz, P., Costello, C.E., Biemann, K.: Matrix-assisted laser desorption ionization mass-spectrometry with 2-(4-hydroxypheny(azo)benzoic acid matrix. J. Am. Soc. Mass Spectrom. 4(5), 399–409 (1993)
Lorkiewicz, P., Yappert, M.C.: 2-(2-Aminoethylamino)-5-nitropyridine as a basic matrix for negative-mode matrix-assisted laser desorption/ionization analysis of phospholipids. J. Mass Spectrom. 44(1), 137–143 (2009)
Mirza, S., Prasada Raju, N., Madhavendra, S.: 5-amino-2-mercapto-1, 3, 4-thiadiazole: a new matrix for the efficient matrix-assisted laser desorption/ionization of neutral carbohydrates. Rapid Commun. Mass Spectrom. 18, 1666–1674 (2004)
Smirnov, I.P., Zhu, X., Taylor, T., Huang, Y., Ross, P., Papayanopoulos, I.A., Martin, S.A., Pappin, D.J.: Suppression of ↦ -cyano-4-hydroxycinnamic acid matrix clusters and reduction of chemical noise in MALDI-TOF mass spectrometry. Anal. Chem. 76(10), 2958–2965 (2004)
Armstrong, D.W., Zhang, L.K., He, L.F., Gross, M.L.: Ionic liquids as matrixes for matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 73(15), 3679–3686 (2001)
Li, Y.L., Gross, M.L.: Ionic-liquid matrices for quantitative analysis by MALDI-TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 15(12), 1833–1837 (2004)
Olson, M.T., Blank, P.S., Sackett, D.L., Yergey, A.L.: Evaluating reproducibility and similarity of mass and intensity data in complex spectra—applications to tubulin. J. Am. Soc. Mass Spectrom. 19(3), 367–374 (2008)
Mcdonnell, L.A., Van Remoortere, A., van Zeijl, R.J.M., Deelder, A.M.: Mass spectrometry image correlation: quantifying colocalization. J. Proteome Res. 7(8), 3619–3627 (2008)
Jiang, W., Qiu, Y., Ni, Y., Su, M., Jia, W., Du, X.: An automated data analysis pipeline for GC-TOF-MS metabonomics studies. J. Proteome Res. 9(11), 5974–5981 (2010)
Heyer, L.J., Kruglyak, S., Yooseph, S.: Exploring expression data: identification and analysis of coexpressed genes. Genome Res. 9(11), 1106–1115 (1999)
Frank, A., Bandeira, N., Shen, Z., Tanner, S., Briggs, S., Smith, R., Pevzner, P.A.: Clustering millions of tandem mass spectra. J. Proteome Res. 7, 113–122 (2008)
Gjukich, M. 2007 T2Extractor. In https://proteomecommons.org/linktracker.jsp?url=http%3A%2F%2Fwww.proteomecommons.org%2Farchive%2F1114637208624%2F. P.C. Administrator, ed: Proteome Comons, Univ. Michigan.
Wolff, J., Sackett, D.L., Knipling, L.: Cation selective promotion of tubulin polymerization by alkali metal chlorides. Protein Sci. 5(10), 2020–2028 (1996)
Sackett, D.L., Knipling, L., Wolff, J.: Isolation of microtubule protein from mammalian brain frozen for extended periods of time. Protein Expr. Purif. 2(5/6), 390–393 (1991)
Sackett, D.L.: Rapid purification of tubulin from tissue and tissue culture cells using solid-phase ion exchange. Anal. Biochem. 228(2), 343–348 (1995)
Woods, A.S., Huang, A.Y., Cotter, R.J., Pasternack, G.R., Pardoll, D.M., Jaffee, E.M.: Simplified high-sensitivity sequencing of a major histocompatibility complex class I-associated immunoreactive peptide using matrix-assisted laser desorption/ionization mass spectrometry. Anal. Biochem. 226(1), 15–25 (1995)
Olson, M.T., Epstein, J.A., Yergey, A.L.: De novo peptide sequencing using exhaustive enumeration of peptide composition. J. Am. Soc. Mass Spectrom. 17(8), 1041–1049 (2006)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Olson, M.T., Epstein, J.A., Sackett, D.L. et al. Production of Reliable MALDI Spectra with Quality Threshold Clustering of Replicates. J. Am. Soc. Mass Spectrom. 22, 969–975 (2011). https://doi.org/10.1007/s13361-011-0097-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-011-0097-9