
Abstract
Hereditary multiple exostoses (HME) also known as multiple osteochondromas represent one of the most frequent bone tumor disorder in humans. Its clinical presentation is characterized by the presence of multiple benign cartilage-capped tumors located most commonly in the juxta-epiphyseal portions of long bones. HME are usually inherited in autosomal dominant manner, however de novo mutations can also occur. In most patients, the disease is caused by alterations in the EXT1 and EXT2 genes. In this study we investigated 33 unrelated Polish probands with the clinical and radiological diagnosis of HME by means of Sanger sequencing and MLPA for all coding exons of EXT1 and EXT2. We demonstrated EXT1 and EXT2 heterozygous mutations in 18 (54.6 %) and ten (30.3 %) probands respectively, which represents a total of 28 (84.9 %) index cases. Sequencing allowed for the detection of causative changes in 26 (78.8 %) probands, whereas MLPA showed intragenic deletions in two (6.1 %) further cases (15 mutations represented novel changes). Our paper is the first report on the results of exhaustive mutational screening of both EXT1/EXT2 genes in Polish patients. The proportion of EXT1/EXT2 mutations in our group was similar to other Caucasian cohorts. However, we found that EXT1 lesions in Polish patients cluster in exons 1 and 2 (55.6 % of all EXT1 mutations). This important finding should lead to the optimization of cost-effectiveness rate of HME diagnostic testing. Therefore, the diagnostic algorithm for HME should include EXT1 sequencing (starting with exons 1–2), followed by EXT2 sequencing, and MLPA/qPCR for intragenic copy number changes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
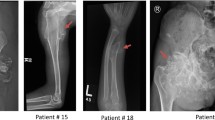
Hereditary multiple exostoses (HME) also referred to as multiple osteochondromas (MO) are one of the most common benign bone tumors with an estimated prevalence rate of 1 per 50,000 in European population (Hennekam 1991; Schmale et al. 1994). The disorder is usually inherited in autosomal dominant manner, however de novo mutations are also known to occur. HME are highly penetrant (close to 100 %) and show significant variability in symptoms expression and the age of onset, which varies from 2 to 15 years (Schmale et al. 1994). Clinical picture of HME involves formation of benign cartilage-capped tumors most frequently located in the juxta-epiphyseal parts of long bones, especially around the knee (femur, tibia), the wrist, the proximal humerus, the proximal fibula, and the ribs. Exostoses can also occur in scapula and pelvis, but neither mandible nor the calvarium are involved (Shapiro et al. 1979; Schmale et al. 1994). Lesions are usually inconspicuous at birth and tend to grow in number and size through childhood and adolescence until the closure of growth plates in puberty. It has been suggested that the formation of exostoses in HME patients is the consequence of a two-hit model in which predisposition for tumor development due to germline mutation and a second somatic “hit” are necessary for the development of exostoses (Mertens et al. 1994). Due to the disruption of growth plates osteochondromas may lead to the various skeletal deformities, such as limb shortening, angular deviation of long bones (especially the ulna), Madelung deformity, movement restrictions, as well as short stature. In addition, exostoses can cause nerve or blood vessel compression, joint limitations, and in some cases (up to 5 %) can transform into malignant tumors such as chondrosarcoma or osteosarcoma (Hennekam 1991; Wicklund et al. 1995).
HME result from the mutations of at least two putative tumor suppressor genes, i.e., EXT1 located on chromosome 8q24.1 and EXT2—located on chromosome 11p11 (Ahn et al. 1995; Stickens et al. 1996; Wuyts et al. 1996). Third region with a hypothetical EXT3 gene was mapped to the chromosome 19p in linkage studies of EXT1/EXT2 negative family, nevertheless causative mutations associated with this locus have not been identified to date (Le Merrer et al. 1994). Mutations in EXT1 gene underlie 56–78 % of HME cases, whereas in EXT2—21–44 % (Jennes et al. 2009). Both EXT1 and EXT2 genes encode for glycotransferase enzymes of endoplasmic reticulum (N-Acetylglucosamine transferase and D-glucuronic acid transferase respectively) involved in the synthesis of heparan sulfate and proteoglycans (Busse et al. 2007). Mutations in these genes have different patterns of concentration. While lesions of EXT1 are scattered throughout the gene, mutations in EXT2 tend to cluster in the first N-terminal part of the protein (Busse et al. 2007; Jennes et al. 2009).
In this study we investigated 33 unrelated Polish index cases with the clinical and radiological diagnosis of HME. We extend the mutational spectrum of the genes and report our diagnostic experience regarding EXT1 and EXT2 screening in Polish patients, who were not represented so far in published molecular studies.
Materials and methods
Patients and clinical information
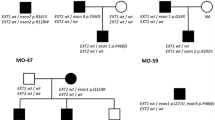
Thirty three unrelated index cases of Polish ethnicity who were clinically and radiographically suspected of HME were recruited for this study. The inclusion criteria involved two or more exostoses diagnosed upon clinical assessment and/or X-ray imaging. Twenty five probands had a positive family history, while eight were sporadic. Blood was collected from all index cases as well as from affected and unaffected available family members. The local ethics committee approved the study and written informed consent was obtained from all subjects or their legal guardians.
Molecular screening
Genomic DNA was isolated from whole blood according to the conventional salting-out method. The coding sequences of both EXT1 and EXT2 genes (GenBank accession number NM_000127 and NM_207122.1), comprised of all coding exons, and the flanking intronic regions were amplified in a set of PCR reactions and directly sequenced by means of dye-terminator chemistry (kit v.3, ABI 3130XL). Sequences of the primers used for amplification and sequencing PCR reactions are given in Table 1 (primers for EXT1 were as described elsewhere by Baasanjav et al. 2010). Multiplex ligation-dependent probe amplification (MLPA) for all exons of the EXT1 and EXT2 was performed with the use of commercial kit P215-B1 per the manufacturer’s protocol (MRC Holland). Data was intra-normalized by dividing the area of each peak by the overall area of the reference probes’ peaks in the probemix. Inter-sample normalization was obtained by comparing the investigated samples to several reference control samples (healthy individuals) run in the same experiment. Relative peak areas ranging from 0.67 to 1.33 were considered normal, below 0.67—deleted, and above 1.33—duplicated (Schouten et al. 2002). DNA of all index cases was screened for both point mutations and intragenic copy number changes involving EXT1 and EXT2 gene, by means of sequencing and MLPA. Next, co-segregation testing was performed in all affected and unaffected family members to check for co-occurrence of the detected mutation with the phenotype. Alternatively, parental studies were done in sporadic cases to confirm a de novo occurrence of the alterations. Detected mutations were referred to Human Gene Mutation Database (HGMD) Professional 2013.4 (HGMD) and Leiden Open Variation Database (LOVD) version 2.0 (Fokkema et al. 2011). Pathogenicity of all identified missense variants was additionally assessed in silico using Mutation Taster 2, Polyphen2, and SIFT software.
Results
We found EXT1 and EXT2 heterozygous mutations in 28 out 33 (84.9 %) unrelated probands from our cohort. In total, we demonstrated 26 different mutational hits, since two of them were recurrent. Eighteen causative alterations (54.6 %) were identified in EXT1, while ten (30.3 %) were shown in EXT2. The remaining five cases (15.1 %) were negative for both EXT1 and EXT2 mutations. DNA sequencing has allowed for the detection of causative changes in 26 (78.8 %) probands, whereas MLPA showed intragenic copy number changes in two (6.1 %) further cases. Out of 28 molecularly confirmed unrelated probands, six cases (21.4 %) occurred due to de novo mutation while 22 (78.6 %) inherited the disease causing variant from an affected parent. According to HGMD® Professional 2013.4 and LOVD v.2.0 databases, out of 26 different mutations demonstrated in this study, 15 alterations were novel, whereas 11 were previously reported elsewhere (HGMD, Fokkema et al. 2011). Nine out of the 15 novel single nucleotide variants (SNVs) were identified in EXT1 and six in EXT2. For the description of the mutations and their reference to literature data, see Table 2. Out of 28 mutations detected by us, 23 (82.1 %) represented inactivating variants (17 frameshift, three nonsense, three splicing mutations). The remaining causative changes were two intragenic deletions (both in EXT2) and three missense substitutions (all in EXT1).
In all sporadic cases, presence of the mutation was excluded in both healthy parents, thus confirming their de novo occurrence in the probands. In familial cases, the identified alterations were checked for co-segregation with the phenotype and were not shown in the unaffected family members. Furthermore, all three missense variants were predicted to be probably damaging in most of the in silico analyses performed by us with the use of Mutation Taster 2, PolyPhen2, and SIFT software (Table 3).
Discussion
Hereditary multiple exostoses is a relatively frequent autosomal dominant bone disorder resulting from heterozygous inactivating mutations of EXT1 and EXT2 genes. Both gene products represent tumor suppressor proteins involved in heparan sulphate (HS) synthesis and cartilage formation. EXT1 and EXT2 act in a hetero-oligomeric complex and catalyze the elongation of HS chains (McCormick et al. 2000; Busse et al. 2007). Mutations in EXT1 or EXT2 are believed to result in reduced level of HS biosynthesis as well as in shortening of HS chains, what disrupts the gradient of morphogens and impair signal transduction in the epiphyseal growth plate in the cartilage. This leads to the loss of cell polarity and once occur in the peripheral part of bone, the cells maintain to proliferate, bring other wild-type cells along and grow into a cartilaginous cap, i.e., osteochondroma (Jones 2011; de Andrea and Hogendoorn 2012).
There are only a few reports on large HME cohorts studied with a comprehensive molecular diagnostic testing including both gene sequencing and quantitative assays, such as MLPA or quantitative PCR (qPCR) for all exons (Porter et al. 2004; Jennes et al. 2009; Ciavarella et al. 2013). Importantly, such studies have never been performed in Polish patients. Thus, our analysis represents the first source of information on molecular cause, frequencies, and the proportion of EXT1 and EXT2 mutations in Polish patients affected by HME. In our study, we demonstrated EXT1 and EXT2 mutations in 28 (84.9 %) out of 33 unrelated probands, which represents a similar diagnostic success rate to previously published reports in which this value varied from 70 to 95 % (Jennes et al. 2009; Ciavarella et al. 2013). Importantly, in EXT1/EXT2 positive Polish patients, EXT1 mutations were found almost twice more frequently than EXT2 mutations (18 vs 10 hits, i.e., 64.3 % vs 35.7 % respectively), which is also in accordance with the literature data (Jennes et al. 2009). The majority of MO causing changes (75–80 %) represent inactivating mutations, i.e., nonsense, frameshift, and splice-site (Jennes et al. 2009). Likewise, as shown in Table 2, 23 out of 28 mutations (82 %) detected in our cohort were also leading to the premature truncation of the protein product. In addition, we found three different EXT1 missense substitutions associated with HME phenotype: p.Y271C, p.R340H, and p.R346G (Fokkema et al. 2011; Raskind et al. 1998; Signori et al. 2007). All missense substitutions were located within exostosin domain of EXT1 and were predicted to be probably damaging to the protein function in computational analyses by means of PolyPhen2, SIFT, and Mutation Taster2 software. An overview of missense mutations, along with their intragenic location, and predictive values of in silico analyses is presented in Table 3.
In keeping with the past reports, EXT1 mutations are usually distributed throughout the entire protein sequence, while EXT2 lesions cluster in the first half of the protein (Jennes et al. 2009). However, in a large Italian cohort described by Ciavarella et al. (2013), 50.1 % of all EXT1 mutations were localized in exons 1 and 2. Our findings based on Polish patients support the hypothesis that there may be an excess of mutations in the first two exons of EXT1. In our cohort, mutation in exons 1 or 2 was identified in ten of 18 (55.6 %) EXT1 mutation carriers, which comprised 30.3 % of our initial cohort (ten probands out of 33). Therefore, we propose that diagnostic screening of HME Polish patients should start with the sequencing of the first two exons of EXT1, followed by sequencing of the rest of the gene. Next, in the case of negative results, we suggest EXT2 sequencing followed by copy number assays for EXT1/EXT2 exons.
To conclude, our paper is the first report that provides the results of exhaustive mutational screening of both HME related genes (EXT1/EXT2) in a Polish cohort. The ratio of EXT1 vs EXT2 gene lesions in our study is in full accordance with the relative mutational frequencies published previously for other Caucasian cohorts. We also expand here the mutational spectrum associated with MOs by describing the 15 novel pathogenic alterations in the EXT1 and EXT2 genes. In addition, although based on a small sample, our study supports the hypothesis that at least in certain groups of patients EXT1 mutations may cluster within the first two exons of the gene. This important finding should influence the diagnostic algorithm, thereby leading to the optimization of cost-effectiveness rate in HME genetic testing. Furthermore, we showed that intragenic EXT1/2 deletions may account for a non-negligible proportion of HME causative mutations. Therefore, we propose that MLPA or qPCR should be implemented into routine molecular diagnostic of the EXT1/2 genes, especially if the sequence analysis detects no pathogenic alteration.
References
Ahn J, Ludecke HJ, Lindow S, Horton WA, Lee B, Wagner MJ, Horsthemke B, Wells DE (1995) Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat Genet 11(2):137–143
Baasanjav S, Jamsheer A, Kolanczyk M, Horn D, Latos T, Hoffmann K, Latos-Bielenska A, Mundlos S (2010) Osteopoikilosis and multiple exostoses caused by novel mutations in LEMD3 and EXT1 genes respectively - coincidence within one family. BMC Med Genet 11:110
Busse M, Feta A, Presto J, Wilén M, Grønning M, Kjellén L, Kusche-Gullberg M (2007) Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J Biol Chem 282(45):32802–32810
Cheung PK, McCormick C, Crawford BE, Esko JD, Tufaro F, Duncan G (2001) Etiological point mutations in the hereditary multiple exostoses gene EXT1: a functional analysis of heparan sulfate polymerase activity. Am J Hum Genet 69(1):55–66
Ciavarella M, Coco M, Baorda F, Stanziale P, Chetta M, Bisceglia L, Palumbo P, Bengala M, Raiteri P, Silengo M, Caldarini C, Facchini R, Lala R, Cavaliere ML, De Brasi D, Pasini B, Zelante L, Guarnieri V, D’Agruma L (2013) 20 novel point mutations and one large deletion in EXT1 and EXT2 genes: report of diagnostic screening in a large Italian cohort of patients affected by hereditary multiple exostosis. Gene 515(2):339–348
de Andrea CE, Hogendoorn PC (2012) Epiphyseal growth plate and secondary peripheral chondrosarcoma: the neighbours matter. J Pathol 226(2):219–228
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT (2011) LOVD v. 2.0: the next generation in gene variant databases. Hum Mutat 32(5):557–563
Francannet C, Cohen-Tanugi A, Le Merrer M, Munnich A, Bonaventure J, Legeai-Mallet L (2001) Genotype-phenotype correlation in hereditary multiple exostoses. J Med Genet 38(7):430–434
Hennekam RC (1991) Hereditary multiple exostoses. J Med Genet 28(4):262–266
HGMD® Professional 2013.4. https://portal.biobase-international.com/. Accessed December 2013
Jennes I, Entius MM, Van Hul E, Parra A, Sangiorgi L, Wuyts W (2008) Mutation screening of EXT1 and EXT2 by denaturing high-performance liquid chromatography, direct sequencing analysis, fluorescence in situ hybridization, and a new multiplex ligation-dependent probe amplification probe set in patients with multiple osteochondromas. J Mol Diagn 10(1):85–92
Jennes I, Pedrini E, Zuntini M, Mordenti M, Balkassmi S, Asteggiano CG, Casey B, Bakker B, Sangiorgi L, Wuyts W (2009) Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum Mutat 30(12):1620–1627
Jones KB (2011) Glycobiology and the growth plate: current concepts in multiple hereditary exostoses. J Pediatr Orthop 31(5):577–586
Le Merrer M, Legeai-Mallet L, Jeannin PM, Horsthemke B, Schinzel A, Plauchu H, Toutain A, Achard F, Munnich A, Maroteaux P (1994) A gene for hereditary multiple exostoses maps to chromosome 19p. Hum Mol Genet 3(5):717–722
McCormick C, Duncan G, Goutsos KT, Tufaro F (2000) The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci U S A 97(2):668–673
Mertens F, Rydholm A, Kreicbergs A, Willén H, Jonsson K, Heim S, Mitelman F, Mandahl N (1994) Loss of chromosome band 8q24 in sporadic osteocartilaginous exostoses. Gene Chromosome Cancer 9(1):8–12
Porter DE, Lonie L, Fraser M, Dobson-Stone C, Porter JR, Monaco AP, Simpson AH (2004) Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype-phenotype study. J Bone Joint Surg Br 86(7):1041–1046
Raskind WH, Conrad EU 3rd, Matsushita M, Wijsman EM, Wells DE, Chapman N, Sandell LJ, Wagner M, Houck J (1998) Evaluation of locus heterogeneity and EXT1 mutations in 34 families with hereditary multiple exostoses. Hum Mutat 11(3):231–239
Sarrión P, Sangorrin A, Urreizti R, Delgado A, Artuch R, Martorell L, Armstrong J, Anton J, Torner F, Vilaseca MA, Nevado J, Lapunzina P, Asteggiano CG, Balcells S, Grinberg D (2013) Mutations in the EXT1 and EXT2 genes in Spanish patients with multiple osteochondromas. Sci Rep 3:1346
Schmale GA, Conrad EU 3rd, Raskind WH (1994) The natural history of hereditary multiple exostoses. J Bone Joint Surg Am 76(7):986–992
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002) Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30(12):e57
Shapiro F, Simon S, Glimcher MJ (1979) Hereditary multiple exostoses. Anthropometric, roentgenographic, and clinical aspects. J Bone Joint Surg Am 61(6A):815–824
Signori E, Massi E, Matera MG, Poscente M, Gravina C, Falcone G, Rosa MA, Rinaldi M, Wuyts W, Seripa D, Dallapiccola B, Fazio VM (2007) A combined analytical approach reveals novel EXT1/2 gene mutations in a large cohort of Italian multiple osteochondromas patients. Gene Chromosome Cancer 46(5):470–477
Stickens D, Clines G, Burbee D, Ramos P, Thomas S, Hogue D, Hecht JT, Lovett M, Evans GA (1996) The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat Genet 14(1):25–32
Vanita V, Sperling K, Sandhu HS, Sandhu PS, Singh JR (2009) Novel EXT1 and EXT2 mutations in hereditary multiple exostoses families of Indian origin. Genet Test Mol Biomark 13(1):43–49
Wicklund CL, Pauli RM, Johnston D, Hecht JT (1995) Natural history study of hereditary multiple exostoses. Am J Med Genet 55(1):43–46
Wuyts W, Van Hul W, Wauters J, Nemtsova M, Reyniers E, Van Hul EV, De Boulle K, de Vries BB, Hendrickx J, Herrygers I, Bossuyt P, Balemans W, Fransen E, Vits L, Coucke P, Nowak NJ, Shows TB, Mallet L, van den Ouweland AM, McGaughran J, Halley DJ, Willems PJ (1996) Positional cloning of a gene involved in hereditary multiple exostoses. Hum Mol Genet 5(10):1547–1557
Wuyts W, Radersma R, Storm K, Vits L (2005) An optimized DHPLC protocol for molecular testing of the EXT1 and EXT2 genes in hereditary multiple osteochondromas. Clin Genet 68(6):542–547
Acknowledgments
We are grateful to the patients and their families for participating in this study. We thank all the referring physicians. This work was supported by a grant from the Polish National Science Centre (UMO-2011-03-D-NZ2-06136) to AJ.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Jamsheer, A., Socha, M., Sowińska-Seidler, A. et al. Mutational screening of EXT1 and EXT2 genes in Polish patients with hereditary multiple exostoses. J Appl Genetics 55, 183–188 (2014). https://doi.org/10.1007/s13353-014-0195-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-014-0195-z