
Abstract
Despite thousands of neuroprotectants demonstrating promise in preclinical trials, a neuroprotective therapeutic has yet to be approved for the treatment of acute brain injuries such as stroke or traumatic brain injury. Developing a more detailed understanding of models and populations demonstrating “neurological resilience” in spite of brain injury can give us important insights into new translational therapies. Resilience is the process of active adaptation to a stressor. In the context of neuroprotection, models of preconditioning and unique animal models of extreme physiology (such as hibernating species) reliably demonstrate resilience in the laboratory setting. In the clinical setting, resilience is observed in young patients and can be found in those with specific genetic polymorphisms. These important examples of resilience can help transform and extend the current neuroprotective framework from simply countering the injurious cascade into one that anticipates, monitors, and optimizes patients’ physiological responses from the time of injury throughout the process of recovery. This review summarizes the underpinnings of key adaptations common to models of resilience and how this understanding can be applied to new neuroprotective approaches.
Similar content being viewed by others


The promise of an acute neuroprotective strategy to minimize brain injuries from stroke, anoxia, or trauma has tantalized neurologists and neuroscientists alike for nearly a century. Despite the plethora of preclinical studies supporting the neuroprotective actions of thousands of therapies, not a single therapy has been approved for use as a neuroprotective agent in patients with brain injuries such as acute ischemic stroke or traumatic brain injury (TBI). Moreover, recent clinical trials have failed to show benefit of mild hypothermia for TBI leading to revisions in the clinical use of the most broadly applied neuroprotective treatment [1]. These translational research and clinical trial challenges have left many in the field frustrated. However, they have led to important new guidelines increasing the rigor of preclinical research methodologies and clinical trial design considerations [2,3,4]. These translational failures have also shifted the focus of neuroprotection research from agents acting on single pathways to broader approaches modulating multiple pathways [5,6,7]. Outcomes for patients with severe brain injuries are still extremely poor, and the development of multifaceted approaches for neuroresilience that support brain cell metabolic recovery, resolve post-injury inflammation, and restore neural circuit functional integrity are long overdue.
Improved Outcomes Through Resilience in Cells and Circuits
Resilience refers to the active process of adapting over time to stressor conditions [8]. The ideal neuroprotective strategy not only counters the molecular and biochemical events following brain injury [9], but also stimulates the necessary adaptations to the post-injury environment to allow for long-lasting neural cell survival and the re-establishment of normal functional connectivity. We review recent steps forward in our understanding of neuronal resilience, the pathophysiology of the most common severe brain injuries, and discuss the next generation of potential therapeutic approaches to stimulate neuroresilience in response to acute brain injuries.
Pathways of Resilience: Lessons from the Laboratory, Nature, and the Clinical Realm
Harnessing the Innate Resilience of the Brain by Conditioning
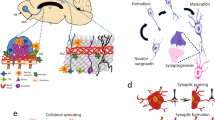
Preconditioning refers to the ability of an exposure to a sublethal cellular or organismal stress to induce cytoprotection or survival to subsequent stressors. Preconditioning was first studied with regard to cardioprotection, and has since been studied extensively in numerous models of tissue injury including stroke, hypoxic-ischemic brain injury, and TBI (for review, see [10, 11]). Preconditioning is neuroprotective in vitro and in vivo following exposure to numerous stimuli including ischemia, hypothermia, and seizures [12,13,14,15]. In addition, transient ischemia in peripheral organs like skeletal muscle can induce remote preconditioning in the brain (for review, see [16]). A robust body of evidence has demonstrated that the adaptive pathways engaged by preconditioning differ fundamentally from mechanisms that simply counter the injury, and demonstrate a genetic reprogramming of the cell’s response to the injury. Pro-survival pathways are stimulated in neurons, glia, microglia, endothelial, and peripheral immune cells via paracrine and autocrine effectors responding to preconditioning stimuli [10, 11]. Together, these adaptations result in improved metabolic efficiency, preservation of the blood-brain barrier, decreased excitotoxicity, induction of anti-apoptotic genes, suppressed inflammation, and enhanced regenerative mechanisms (see Fig. 1).

Paracrine factors involved in the resilience pathways of innate and preconditioning models of neuroprotection. Preconditioning and innate ischemia tolerance are mediated by complex autocrine and paracrine interactions between neurons, astrocytes, microglia, and endothelial cells. Together, these interactions contribute to resilience by: decreasing neuronal metabolic demands and apoptosis; improving astrocytic glutamate handling and blood-brain barrier stabilization; and decreasing microglial activation and promoting synaptogenesis and structural plasticity. Abbreviations: BDNF: brain-derived neurotrophic factor; EPO: erythropoietin; HIF: hypoxia-inducible factor; HO: heme oxygenase; IL: interleukin; MMP: matrix metallopeptidase; TLR: toll-like receptor; TNF: tumor necrosis factor; VEGF: vascular endothelial growth factor
The effects of ischemic preconditioning on global regulators of cellular stress response pathways such as transcription factors, protein kinases, or epigenetic modifiers are particularly intriguing from a translational perspective, as these are ideal neuroprotective agent candidates to effectively stimulate diverse and coordinated pro-survival pathways (see Fig. 2). Hypoxia-inducible factor (HIF)-1α, a transcription factor regulating dozens of target genes such as vascular endothelial growth factor (VEGF), erythropoietin (EPO), and nuclear-encoded mitochondrial proteins like Cox (cytochrome c oxidase)-4, is one such candidate [17, 18]. Ischemic preconditioning increases HIF1α by decreasing prolyl-hydroxylase-2, the constitutively active HIF1α degradation enzyme [19]. By regulating the transcription of VEGF and EPO, HIF1α enhances oxygen delivery to tissues and stabilizes endothelial-astrocytic interactions supporting BBB integrity. HIF1α also improves cellular metabolic efficiency and reduces reactive oxygen species (ROS) by regulating the expression of specific oxidative phosphorylation (OXPHOS) enzyme subunits and stimulating mitochondrial autophagy (the regulated removal of unnecessary or damaged cellular components) [17, 20,21,22]. Although HIF1α clearly has beneficial action with regard to recovery from ischemic injury, it mediates some detrimental effects of hypoxia in a temporally dependent manner as well [23]. This suggests that like many other effectors of preconditioning, a complex interplay exists between its temporal and cell type–specific expression in response to ischemia, which will have to be carefully considered in translating therapies to acute brain injuries.

Schematic model of key cellular resilience pathways in innate and preconditioning models of neuroprotection. Preconditioned or ischemia-tolerant organisms like the Arctic ground squirrel possess increased levels of (1) key transcription factors including NRF2, HIF1⍺, CREB, and NFKB which increase the expression of (2) numerous target genes coordinating a protective response to (subsequent) ischemia. These transcriptional responses decrease ischemia-induced (3) intracellular calcium via sarcoendoplasmic reticulum calcium transport ATPase. Increased mitochondrial metabolic efficiency is also mediated by (4) SUMOylation and (5) PKCε post-translational modifications which converge to (6) increase ischemic ATP production and reduce ROS, resulting in cytoprotection. Abbreviations: BDNF: brain-derived neurotrophic factor; Cox: cytochrome c oxidase; CREB: cyclic AMP response element-binding protein; EPO: erythropoietin; ER: endoplasmic reticulum; HIF: hypoxia-inducible factor; HO: heme oxygenase; iNOS: inducible nitric oxide synthase; MPTP: mitochondrial permeability transition pore; NFKB: nuclear factor light-kappa-chain-enhancer of activated B cells; Nrf2: Nuclear factor erythroid 2-related factor 2; PKC: protein kinase C; ROS: reactive oxygen species; SENP: sentrin-specific protease; SOD: supraoxide dismutase; SUMO: small ubiquitin-like modifier; VEGF: vascular endothelial growth factor
Another key effector of preconditioning acting on numerous cellular stress response pathways is protein kinase C (PKC)-ε. PKCε is one of 12 known isozymes of the PKC family, which are multifunctional protein kinases that phosphorylate serine and threonine residues in many target proteins. PKCε is rapidly activated by ischemia-induced increases in intracellular calcium and diacylglycerol and is required for hypoxia preconditioning in hippocampal slices [24]. PKCε functions to protect mitochondria from ischemia by phosphorylating numerous other key regulators of cellular energy homeostasis including Akt, MAPK, and AMPK. It is also translocated to the mitochondria following preconditioning where it phosphorylates mitochondrial KATP channels, which decreases mitochondrial ROS and apoptosis [25]. Interestingly, brain-derived neurotrophic factor (BDNF), a major regulator of synaptic structure, function, and plasticity, is also upregulated following preconditioning [26, 27] and acts downstream of PKCε to induce neuroprotective changes [28, 29].
Epigenetic modifications following preconditioning are also critical to its neuroprotective effects (for review, see [30]. The small ubiquitin-like modifier (SUMO) family of proteins are key post-translational modifications that affect gene expression by altering recruitment of histone deacetylases. SUMOylation modulates protein function by affecting proteasomal degradation or sites of interactions with other proteins and substrates. SUMOylation is stimulated by ischemia and ischemic preconditioning in vitro and in vivo [31,32,33,34,35]. Conjugation of SUMO is rapid after ischemia and key to its neuroprotective effects [36, 37]. Thousands of proteins are targets of SUMOylation, and the specific subsets mediating preconditioning and its protective effects are not known [38]. However, global inhibitors of SUMO-specific proteases (SENPs), which are responsible for de-SUMOylation of proteins, facilitate neuroprotection [39]. Dynamic regulation of SUMOylation is also important in mitochondrial dynamics, protein quality control, synaptogenesis, and synaptic plasticity, suggesting multifaceted mechanisms of its neuroprotective effects [40,41,42].
Ischemic preconditioning also improves recovery after brain injuries by suppressing inflammatory responses. Inflammatory mediators such as lipopolysaccharide, tumor necrosis factor (TNF)-α, and interleukin-1 can induce preconditioning independently of ischemic stimuli [43]. Toll-like receptor (TLR)-4 and other TLRs, which are highly expressed in microglia, are also essential for ischemic preconditioning, possibly via TNFα action [44,45,46]. Astrocytic-microglial cross-talk may be particularly relevant to reducing microglial activation and leukocyte infiltration after ischemic preconditioning. Increased astrocytic heme oxygenase (HO)-1 and interleukin-10 expression reduces microglial ROS and activation, while microglial cytokines help induce a neuroprotective astrocytic phenotype via downregulation of the purinergic receptor P2Y1 [47,48,49].
Clinical evidence of preconditioning for neuroprotection has not been demonstrated prospectively; however, several intriguing retrospective analyses demonstrate that stroke severity is reduced in patients where stroke was preceded by transient ischemic attack [50]. In addition to the paracrine factors described above, clinical preconditioning may lead to neuro-cardiovascular adaptations providing increased cerebral blood flow such as cerebral vasodilation and increased cerebral perfusion pressure. Conditioning stimulates the release of protective circulating chemokines, EPO, and other factors that reduce neural damage and inflammation following ischemia [11].
Resilient Adaptations of Hibernating Mammals
Hibernation is a survival strategy used by animals living in harsh environments characterized by a prolonged state of energy conservation. During hibernation in small mammals, there is a profound reduction of metabolic rate and core temperature to match the 90% reduction of cerebral perfusion [51]. Intriguingly, in Arctic ground squirrels (AGS) and many other small mammal hibernators, late winter hibernation is interrupted every 10–21 days by interbout arousal (IBA) episodes. During episodes of IBA, cerebral blood flow fluctuates rapidly, yet AGS suffer no ischemic or reperfusion injuries [52]. This phenotype is also recapitulated in vitro, as hippocampal slices from euthermic AGS demonstrate markedly improved tolerance to oxygen glucose deprivation compared to rat hippocampal slices [53]. Interestingly, this study demonstrated that tolerance to oxygen glucose deprivation in AGS occurs independent of hibernation season and persists in conditions mimicking ischemia-reperfusion such as following depletion of ATP, glutamate excitotoxicity, and acidosis. As in hypoxia preconditioning, reduced release of ROS and reactive nitrogen species in response to oxygen glucose deprivation are key factors underlying reduced cell death in AGS [54]. While the bioenergetic feats of AGS hibernation are notable, perhaps even more astounding is the rapid dendritic regrowth and synaptic rebuilding accompanying each IBA [55]. The molecular underpinnings of this remarkable metabolic and structural plasticity are a topic of active investigation. Recent studies in hibernating ground squirrel neurons derived from induced pluripotent stem cells have found significant adaptations in cytoskeletal and lysosomal pathways governing responses to hypothermia and cellular stresses [56]. Analysis of hibernating mammalian genomes has identified numerous adaptations in pathways regulating intracellular calcium homeostasis, OXPHOS machinery, and antioxidant defense systems [57]. Transcriptomic and proteomic studies have found that the neural adaptations underlying hibernation parallel the adaptations of preconditioning in many ways [30, 58]. These overlapping mechanisms include the ability to rapidly downregulate energy-consuming protein synthesis, ion channel permeability, inflammatory responses, oxidative stress, and cell death pathways.
Several hibernating species have been studied in non-hibernating conditions in models of ischemia in the laboratory. In general, hibernating species survive hypoxia and ischemia better than non-hibernating species [59]. This is related to ischemic metabolic responses mirroring those hibernating animals’ encounter upon entering hibernation as well as genetic differences conferring innate ischemia tolerance [60]. Interestingly, hibernating species demonstrate elevated levels of effectors of preconditioning including HIF1, inducible nitric oxide synthase (iNOS), PKCε, SUMOylation, and antioxidants such as supraoxide dismutase (SOD) and HO1 [61,62,63,64,65]. Hibernating animals also exhibit numerous physiological adaptations including the use of alternative fuels for cerebral metabolism (ketones), elevated antioxidants, reduced inflammatory responses, and enhanced oxygen delivery [66].
Taken together, the research in this field demonstrates that hibernating species are robust examples of ischemic tolerance. This is underscored by endogenous cellular protection from the metabolic stress of ischemia, dramatic flexibility in synaptic structure and function, and physiologic changes in metabolism and immune responses. Modern genomic and metabolomic techniques are producing important insights into the molecular and physiological underpinnings of this resilience as well as how they could be applied to treating patients with brain injuries. The failure of TBI and stroke neuroprotection trials focusing on single agents suggests that new approaches mimicking the coordinated cellular and physiological responses of resilient species may lead to improved outcomes.
The Resilience Within—Age and Genetic Variation Influence Recovery from Brain Injury
Clinicians are familiar with the sometimes marked improvements young patients can make following devastating brain injuries. This is in stark contrast to the often-limited recovery observed in older patients. This anecdotal experience is backed up by epidemiological evidence, as the greatest improvements in early functional recovery are age-related, with patients in early adolescence making the best functional gains [67]. Preclinical studies in rodents and pigs demonstrate greater lesions in aged animals after controlled cortical impact (CCI; [68, 69]). Numerous studies suggest that the pro-survival and recovery phenotype of young animals is due to improved mitochondrial metabolism, decreased oxidative stress, enhanced BBB and neurovascular integrity following injury, and a faster resolution of post-injury inflammation [70,71,72,73,74,75,76]. Recovery from stroke in young animals appears to be particularly accelerated due to decreased microglial activation and systemic inflammation [77]. Interestingly, Ritzel and colleagues performed transplants of young bone marrow in older mice and vice versa. They found that bone marrow rejuvenation reduces the severity of acute behavioral deficits after stroke, while bone marrow senescence increases neutrophilic infiltration of infarcts. They also examined human post-mortem stroke tissues and found that age correlated with neutrophil counts, matrix metallopeptidase (MMP)-9 expression, and the presence of microhemorrhages [78].
Examining the effects of single nucleotide polymorphisms (SNPs) on brain injury and recovery can also provide insights into pathways of resilience. Although there are few high-quality and sufficiently powered studies in this field, there are small studies with intriguing findings in patients with TBI and stroke that will require further validation. In the largest genome-wide association study of stroke recovery with 6,165 patients, Soderholm and colleagues found only one significant gene locus (rs1842681), which corresponds to an intronic sequence thought to regulate the expression of a regulatory subunit of protein phosphatase 1 (PP1; [79]). Although the specific function of this regulatory subunit is not yet known, PP1 is part of pathway that plays a prominent role in cellular responses to stress and preconditioning [80,81,82]. Numerous SNPs have been associated with recovery in TBI (for review [83, 84]. While larger studies are necessary to validate many of the identified loci, there is a notable overlap with the resilience pathways described above including genes involved in BBB and endothelial integrity (several cytokines and aquaporin), metabolic function (3 mitochondrial genes), and neurotrophic factors (BDNF).
Taken together, these investigations spanning the bench-to-bedside continuum point to the importance of metabolic efficiency, anti-apoptotic genes, BBB integrity, immune responses, and synaptic plasticity/regeneration in conferring resilience to brain injuries. These pathways have been the focus of intense research, but additional mechanistic and translational work remains to fully understand the key interacting factors in these pathways, the optimal treatment approaches, and the timing of treatment. The above diverse examples of resilience provide a blueprint for beginning to understanding which patient variables require monitoring and titration in the design of future neuroprotective strategies. In particular, as these mechanisms co-exist in animal models of ischemic tolerance, multifunctional therapies targeting multiple resilience pathways may be more likely to confer functional benefits to patients. Below we discuss pathophysiological insights and potential neuroresilience targets for TBI and stroke. Although these two entities are the most common brain injuries and have been extensively studied in preclinical and clinical paradigms, the bioenergetic processes underlying neural injury as well as the inflammatory and repair processes accompanying recovery in these diseases are also relevant to many other acute central nervous system injuries including intracerebral hemorrhage, vasospasm after subarachnoid hemorrhage, seizures, and spinal cord injury.
Traumatic Brain Injury
Traumatic brain injury is a major cause of death and disability worldwide. In the USA alone, nearly 3 million people seek out care for TBI yearly with direct medical costs estimated to be over $11 billion [85]. In the USA and Europe, the greatest incidence is in those < 25 and > 65 years of age [86, 87]. A key challenge in promoting neural resilience following TBI is the heterogeneity in the mechanisms of injury and underlying physiological and cellular processes accompanying the primary and secondary brain injuries. Primary injuries (hematomas, contusions, axonal stretching) are caused by the kinetic energy from external physical forces (blows, acceleration/deceleration, and rotation) at the time of impact. Secondary injuries can result from several physiological processes including increased intracranial hypertension, hypotension, hypoxia, hypo/hyperglycemia, hyperthermia, seizures, or cortical spreading depressions and exacerbate the primary injury in the acute and subacute period following the initial injury. The cellular and molecular pathways underlying this myriad of secondary injuries are complex, and include ischemia, cytotoxic and/or vasogenic brain edema, dysregulation of the BBB, and dysfunctional autoregulatory responses [88].
Treatment Approach
For patients with severe TBI, urgent neurosurgical intervention is often required for management of elevated intracranial pressure (ICP). After acute stabilization, the central focus of TBI patient care is to limit secondary brain injury by preserving cerebral blood flow to ischemic tissue, reducing mass effect due to cytotoxic edema, preventing seizure-related metabolic crises, and maintaining physiological homeostasis with regard to cerebral perfusion pressure (CPP), oxygenation, glucose levels, temperature, and other physiologic variables as possible. Although the clinical neurological examination remains the most important indicator of patient recovery, in the Neuro-ICU setting, clinicians are increasingly incorporating a host of additional adjunctive neurological monitoring information to guide treatments in real-time including pupillometry, ICP, cerebral perfusion pressure (CPP), brain tissue oxygenation, jugular venous oximetry, electroencephalogram (EEG), or cerebral microdialysis. Simultaneous use of several monitoring modalities allows clinicians to cross-validate the need for and effect of physiological therapies such as increasing CPP, decreasing ICP, or improving oxygenation [89]. Given the amount of data generated with multimodality monitoring, it is expected that implementation of robust neuro-informatics and deep learning algorithms will provide valuable new insights into patient-specific physiology underlying secondary injuries and greatly assist the development of new therapeutic approaches [90, 91]. Indeed, clinical decision support algorithms based on multimodality monitoring are being developed and the efficacy of some of these is being tested in clinical trials [92].
Recent Neuroprotectant Trials
Over the last several decades, a number of therapeutic strategies have been explored in TBI, but no neuroprotective agent or treatment has been confirmed in large randomized trials as being effective in the clinical setting (for review, see [93]. Several high-profile trials have recently been completed for the early administration of progesterone after TBI as well as the use of mild hypothermia. Progesterone is highly efficacious in preclinical models of TBI, showing benefit in many of the resilience pathways outlined above [94]. It also demonstrated safety and possible efficacy in two phase II human trials; however, it failed to show benefit in larger phase III trials [95, 96]. The application of systemic hypothermia is also supported by an extensive preclinical literature. Its mechanisms are also multifaceted and act upon key resilience pathways. However, several recent notable trials of mild hypothermia and targeted temperature management also found that induction of hypothermia to 33 °C either as a prophylactic strategy or in cases of elevated ICP does not lead to improved outcomes after TBI [1, 97, 98]. The failure of these two highly studied neuroprotectants has been the subject of numerous editorials, and overall reinforces the importance of considering sex difference, patient comorbidities, and systemic complications in preclinical trials as non-neurological complications were likely key factors in the failure of both treatments to improve patient outcome [99, 100]. For example, in ProTECT III, progesterone treatment was associated with a significantly poorer outcome in women and higher rates of non-neurological death. Additionally, in the Eurotherm3235 trial of hypothermia in patients with TBI and elevated ICP, treatment was associated with an increased risk of death (hazard ratio 1.8), which may have been related to an increased mortality effect of multiple organ failure in hypothermia patients [101].
New Pathophysiological Insights and Opportunities for Progress
The resilience framework suggests four interconnected but distinct biological processes that are opportunities for intervention in TBI, including (i) subacute intermittent metabolic crises, (ii) BBB and neurovascular dysfunction, (iii) post-injury inflammation, and (iv) loss of neural circuit integrity. Anticipating and enhancing endogenous adaptations to these deleterious pathways following TBI is key to improving functional outcomes. These processes begin immediately after TBI but peak and resolve at different time courses (Fig. 3). Further understanding the temporal interactions between key mediators of these pathways and neuroprotective treatments will be critical for establishing robust neuroresilience therapies for future clinical trials.

Key cellular, immunologic, and endogenous repair mechanisms following brain injury illustrate therapeutic opportunities. A more detailed characterization of the temporal events accompanying specific neural injuries such as TBI and stroke will help guide the development of more specific neuroprotective therapies to optimize recovery. Treatment protocols based on biomarkers related to the phase of the injury and treatment response hold the promise of maximizing recovery for individual patients. The temporal events and figure panels were derived and adapted from prior works [102,103,104]. Abbreviations: ROS: reactive oxygen species; CSD: cortical spreading depression; PID: peri-infarct depolarization; DAMP: damage-associated molecular pattern; CPP: cerebral perfusion pressure
Metabolic Therapies
Studies combining EEG obtained from depth electrodes with cerebral microdialysis acutely after brain injury have advanced the concept that TBI patients are susceptible to metabolic “crises,” arising from regional ischemia, seizures, inflammation, or other injury-related processes causing intrinsic neuronal metabolic dysfunction [105]. This is corroborated by animal studies finding an acute rise in the demands of mitochondrial electron transport following TBI [106], which results in the enhanced production of ROS. The metabolic failure and accumulation of ROS lead to mitochondrial damage and subsequent neuronal apoptosis [107]. Mild hypothermia has been widely used to reduce metabolic demands of the brain; however, as discussed above, recent evidence of significant systemic complications now precludes its standard clinical use in favor of targeted temperature management to avoid fever [1]. Thus, the goal of the next generation of neuronal metabolism-focused therapies is reducing metabolic crises without systemic complications. Although devices for selective brain cooling are under development [108], identification of the molecular effectors of hypothermia may be a way to deliver its neuroprotective effects while foregoing its systemic cardiovascular and other complications. Numerous mediators of the effects of hypothermia have been identified including RNA-binding motif protein-3 and PKC isoforms, but further pharmacologic and preclinical development is necessary before these therapies will be clinically viable [109]. Physiologic strategies other than hypothermia to reduce metabolic crises will soon enter clinical trials, and include optimizing brain tissue oxygenation monitoring to guide clinical TBI care (BOOST3, phase III) and hyperbaric oxygen treatment (HOBIT, phase II).
The use of alternative fuels to support ATP synthesis in the subacute period following TBI is also an attractive target to increase metabolic resilience. The use of substrates that drive complex II metabolism (succinate dehydrogenase) has been highlighted as a means of bypassing post-injury pyruvate dehydrogenase and complex I dysfunction [110]. An intriguing “within-subjects” study of 9 patients with TBI receiving succinate via an intracranial microdialysis catheter found that its administration compared to a preceding vehicle administration led to decreased glutamate and lactate:pyruvate ratio [111]. Preclinical studies have found benefit in other fuels such as acetyl l-carnitine, increased free fatty acids (via fasting), and ketosis [112, 113]. A small pilot study of l-carnitine failed to show benefit in mortality for TBI patients; however, there was a modest improvement in behavioral outcomes [114]. Induction of ketosis has not yet been examined in clinical trials, but a safety and feasibility study is currently recruiting patients (KETI: NCT03982602).
A major consequence of ROS is mitochondrial damage, which can then activate apoptotic pathways. Protecting mitochondria with antioxidant therapy such as PEG-conjugated SOD and tirilazad [115, 116] has not proven successful in prior TBI trials. While these first-generation therapies were hypothesized to antagonize the free radicals produced by injury, newer therapies act on transcription factors regulating the endogenous production of ROS such as Nrf2. This broader and more potent mechanism may lead to a benefit, particularly if coupled to other protective treatments [117]. Another more recently studied mitochondrial adaptation found in hibernating species and preconditioning is enhanced autophagocytic clearance of damaged mitochondria. Damaged mitochondria increase ROS, and stimulating autophagocytic clearance of mitochondria attenuates neuronal death and functional deficits in an animal model of CCI [118]. Thus, drugs improving autophagy efficiency may improve outcome after brain injury.
Maintaining BBB and Neurovascular Integrity
Ample evidence in patients and animal models of TBI strongly implicates the role of BBB integrity and stability in modulating outcome after TBI [70, 119]. Disruption of the BBB, which is composed of tightly connected endothelial cells, neurons, astrocytes, and pericytes, displays a biphasic peak after TBI. The first occurs at 4–6 h after TBI, and the second is 2–3 days after injury [120]. These periods of increased BBB permeability are mediated by inflammatory and immune activation, although the mechanistic details are still poorly understood. BBB disruption can persist for several months or even years after TBI and may provide a mechanistic link between incident trauma and development of post-concussive symptoms, post-traumatic epilepsy, or progression of neurodegeneration [121]. Loss of BBB integrity following TBI allows for an influx of toxic molecules including thrombin, fibrinogen, and glutamate which further exacerbates neural injury [122].
Maintaining the BBB in animal models of TBI by stabilizing endothelial cell interactions or enhancing pericyte function leads to improved outcomes [123]. Indeed emerging evidence suggests that loss of pericyte function may be an important factor not only in acute brain injury, but also in neurodegenerative disease where pericyte dysfunction impairs cerebral blood flow and leads to metabolic stress and ultimately neuronal death [124]. Interestingly, pericyte death during ischemic phenomena (such as cortical spreading depressions) leads to microcirculatory constriction and further impairs blood flow suggesting that targeting pericyte function and survival could broadly reduce secondary neural injury and improve neuronal survival [125]. While pericytes are not well studied in innate or acquired models of ischemia tolerance, there is emerging evidence of their vulnerability and compromised function in aging, suggesting that pericytes may contribute to resilience observed in younger patients [126].
Reducing Pathogenic Inflammation
The TBI inflammatory cascade is complex with multiple cellular events triggering activation of microglia, recruitment of macrophages, and infiltration of leukocytes [102]. This immunologic mobilization has both harmful and beneficial actions. M1 macrophage/microglial polarization is responsible for pro-inflammatory signaling that damages neurons and further exacerbates local edema, while M2 macrophage/microglial polarization is critical to the resolution of injury and initiation of recovery [127]. Although macrophage/microglial polarization is beginning to be understood as less of a dichotomy, numerous immune-modulating agents are in early clinical trials and aiming to bias M2 in hopes of resolving inflammation and stimulating recovery. However, the heterogeneity of TBI and individual differences in immune responses, modulating post-injury inflammation, may require new biomarkers to titrate patient-specific therapies and this important area of research is still in its infancy [103].
Cell-based therapies including the use of mesenchymal stem cells (MSCs) have important immune-dampening effects following brain injury. Contrary to initial hypotheses, MSCs and other stem cells appear to exert most of their beneficial effects by modulating inflammation, improving the chemokine and growth factor milieu, and enhancing neurogenesis [128]. There is mixed evidence with regard to the ability of transplanted stem cells to reach sites of neural injury, differentiate into neurons or glia, and develop functionally important connections [129]. However, there is ample evidence that stem cells can suppress damage-promoting inflammatory mediators such as TNFα, CCL2, and other chemokines and thereby reducing the detrimental effects of leukocytes, neutrophils, and microglia after TBI. Stem cells also stimulate endogenous recovery mechanisms by providing trophic support to surviving cells [130]. Given the predominantly anti-inflammatory paracrine actions of MSCs and other stem cells, investigators have begun to study the effects of MSC-derived exosomes on recovery from TBI [131,132,133]. Exosomes are small endosomal vesicles released by all cell types and contain molecular cargoes including proteins, lipids, mRNAs, and microRNAs. Exosomes have been shown to regulate a wide variety of biological process including neuroinflammation. Using stem cell–derived exosomes is an attractive alternative to MSCs, as it would circumvent issues related to preservation, storage, transfer, and immunogenicity of stem cells. Given these benefits and the initial preclinical data supporting beneficial actions of MSC-derived exosomes, further follow-up for the use of MSC-derived exosomes as an immune-modulating therapy is merited.
Maintaining Functional Connectivity
The plasticity in regenerating functional synapses observed in hibernating animals as well as relationship between functional outcome and preserved resting-state connectivity in patients points to the importance encouraging circuit-level resilience in TBI patients [134, 135]. Diffuse axonal injuries (DAIs) can lead to large-scale changes in circuit connectivity [136]. New models of brain circuit functional connectivity explain how DAI leads to inefficient neural communication and potentially explains the proportion of neurological deficits seen after DAI, which is often out of proportion to imaging findings [136, 137]. Recent studies point to the role of tau, which is highly expressed in axons, in mediating the pathological changes in neural circuits following DAI [138]. Mechanical stress during TBI has been shown to lead to axonal fractures, which contributes to hyperphosphorylation and aggregation of tau, a microtubule protein. Tau aggregation leads to a cascade of cellular events resulting in axonal degeneration and cell death, potentially linking changes in resting-state connectivity observed in brain injuries to a pathophysiological mechanism. Interestingly, in AGS and other hibernating animals, hyper-phosphorylated tau formed during hibernation and rapidly and fully reversed upon arousal [139]. Although no clinical trials have examined whether targeting tau can improve connectivity and outcome in TBI patients, antibodies directed towards the pathogenic variant of tau are being developed to test this hypothesis [140, 141].
Acute Ischemic Stroke
In 2017, there were approximately 795,000 ischemic strokes in the USA, making it a leading cause of long-term disability and the 5th leading cause of death nationwide [142]. The yearly total costs associated with stroke are over $30 billion. Fundamentally, one of the biggest challenges in ischemic stroke care revolves around the concept of stopping the progression of vulnerable, yet salvageable ischemic tissue (i.e., the penumbra) from becoming permanently infarcted tissue (i.e., the core). This makes time of the essence in treating patients with strokes. Stroke outcomes are drastically improved in those receiving acute stroke treatments and re-establishing blood flow at early time points. Conversely, patients presenting later or with poor collateral circulation suffer larger infarcts and worse outcomes [143].
Treatment Approach
The treatment paradigm in acute ischemic stroke management is centered upon ensuring reperfusion to the injured tissue, thereby, minimizing primary injury. The prominent collateral circulation of the brain compensates in a limited way for the reduced blood flow and can extend windows of opportunity for treatment in some patients. The ischemic injury resulting from stroke triggers secondary injury and immune pathways similar to TBI, and similar to TBI no modulators of these pathways have been approved for improving recovery from stroke. The first stroke therapy, IV tissue plasminogen activator, was approved in 1995, and although subsequent studies have expanded the treatment window [144,145,146], despite only 30% of patients achieving successful reperfusion, it remained the standard of care for 20 years [147]. Since 2015, endovascular thrombectomy (EVT) has revolutionized the treatment of acute stroke, as EVT in patients with large-vessel occlusions results in > 80% successful reperfusion, and markedly improved clinical outcomes [148,149,150,151,152]. In a pooled meta-analysis of 1,287 patients, 46% of patients receiving mechanical embolectomy achieved functional independence at 90 days, compared to 26.5% in the control arm—a number needed to treat of only five [153]. The recent publications of DAWN and DEFUSE-III have further expanded the treatment windows to as far as 24 h [154, 155]. Taken together, these trials advanced the notion that given timely reperfusion, collateral circulation and other endogenous compensatory mechanisms can often salvage the ischemic penumbra from becoming infarcted.
Recent Neuroprotectant Trials
There have been hundreds of clinical trials of neuroprotectant agents in acute ischemic stroke, but none have demonstrated efficacy in a large-scale clinical trial. Numerous articles have been devoted to the methodological shortcomings of these clinical trials and failures of translational science (for reviews, see [9, 156, 157]. However, it is worth highlighting that with the advent of EVT and its high rate of recanalization, it is now possible to administer neuroprotectant therapy quickly and have it reach penumbral tissues effectively as part of routine clinical treatment with EVT. Thus, EVT has reignited enthusiasm for clinical trials of neuroprotection in stroke patients [158, 159]. The success of EVT also has important implications for research in preclinical models of stroke. Previously, research steering committees and investigators advocated for the use of permanent middle cerebral artery occlusion (MCAO) models of stroke given the historically low rates of recanalization for most stroke patients; however, in the era of EVT, further preclinical development of drugs utilizing the transient MCAO model may yield more clinically translatable findings in a population that stands to benefit from drug treatments [160, 161]. As such, although relatively few large clinical trials of pharmacologic neuroprotective agents have been performed in the last several years, the coming decade will bring many new neuroprotection in stroke patients.
One of the most prominent and innovative recent neuroprotectant trials engaged prehospital responders to deliver magnesium sulfate ultra early (FAST-MAG) in patients with suspected stroke [162]. Magnesium is thought to exert its neuroprotective effects by antagonizing NMDA signaling and limiting excitotoxicity. Compelling preclinical data in stroke and other models of brain injury also supports the use of magnesium [163]. The FAST-MAG trial is unique because first responders delivered treatment within a median time of 45 min from symptom onset, addressing concerns that early neuroprotective trials had failed due to significant delays in treatment times. Unfortunately, prehospital initiation of magnesium sulfate did not improve disability outcomes or mortality at 90 days. Although magnesium therapy was not effective, the trial was extremely instructive as an example of the capabilities and limitations of delivering a neuroprotective agent in the prehospital setting.
Clinically, induced or permissive hypertension is often utilized to maintain higher cerebral perfusion pressures prior to reperfusion in patients with stroke. Although the evidence for this is poor [164], it was hypothesized that albumin may have neuroprotective effects by acting as an expander of intravascular volume to preserve penumbral blood flow. However, a phase III study of albumin administration in acute stroke was stopped early due to futility, as after 841 patients were enrolled there was no difference in the percentage achieving a good neurologic outcome at 90 days [165]. Patients receiving albumin were also more likely to experience pulmonary edema and intracranial hemorrhage. Given the successes of embolectomy, revisiting prior failed agents such as magnesium and albumin specifically in patients with large penumbral volumes is of great interest.
New Pathophysiological Insights and Opportunities for Progress
Metabolic Therapies to Freeze the Penumbra
The success of mechanical reperfusion therapies in improving patient outcomes has reinvigorated the neuroprotection field to search for therapies capable of halting core infarct growth. A key factor preventing greater numbers of stroke patients from being treated with and benefitting from EVT is the size of core infarct at presentation. Patients with a large core infarct are not eligible for EVT, and for every 10 cc increase in pre-treatment core, the odds of a favorable outcome decrease by 20–30% after embolectomy [166]. As such, neuroprotection strategies designed to “freeze the penumbra” are highly likely to improve treated patient outcome as well as increase the number of patients that will qualify for reperfusion therapies.
Hypothermia has been well established as a cytoprotective agent, with pleotropic effects on numerous protective pathways. In the acute phase, cooling decreases the accumulation of excitotoxic glutamate, reduces lactate formation and cellular acidosis, preserves ATP, and reduces metabolic demand in response to ischemia [109]. In the subacute phase, hypothermia has been shown to prevent apoptotic cell death, inhibit inflammatory cell infiltration, limit the production of ROS, and maintain BBB integrity. In ischemic stroke, impaired blood flow leads to damage of the tight-junction proteins, basement membranes, and astrocytes, making the BBB prone to injury [167]. Upon reperfusion (i.e., after tPA or EVT), injury can result in the form edema/hyperemia and hemorrhagic transformation. Hypothermia may help prevent such damage by inhibiting the proteolytic activity of MMPs, limiting the degradation of tight junctions, and ensuring the integrity of the basement membrane [168]. The constellation of these protective mechanisms translated into infarct volume reductions of up to 40%, as seen in animal models with MCAO [169]. Key findings from meta-analyses highlighted the importance of inducing hypothermia early in the time course of ischemia and targeting the lowest tolerable temperatures—with the caveat of more adverse effects. These preclinical studies ultimately led to the first clinical trials investigating the safety of hypothermia in ischemic stroke patients [170, 171], which demonstrated feasibility, but with notable high rates of pneumonia. The application of therapeutic hypothermia has since been applied to patients receiving IV-tPA [172], but with limited benefit. To date, there have been no randomized controlled trials demonstrating the efficacy of hypothermia in ischemic stroke, and given the recent trials in TBI demonstrated a high rate of systemic complications; it is likely that clinical trials will focus on selective brain cooling rather than whole-body hypothermia. With the advent of EVT, the notion of using hypothermia as an adjunctive therapy to not only preserve penumbra, but also reduce secondary injury from BBB breakdown is plausible and appealing. In a phase I study in 2013, intravascular cooling to a targeted temperature of 33 °C was safely achieved in 20 patients immediately after successful reperfusion with mechanical thrombectomy. Although limited by a small samples size, patients exhibited lower rates of BBB breakdown and hemorrhagic transformation compared to historical controls [173]. A similar study compared two cohorts from two separate centers, one that performed hypothermia after intra-arterial therapy and another that did not, and found reductions in edema and hemorrhagic transformation among the targeted temperature group [174]. Selective intra-arterial cooling directly to the ischemic bed after endovascular recanalization has also been documented [175]. While conclusions cannot be drawn from these small studies, it highlights the prospect of selective hypothermia as an adjuvant tool in a new age of intra-arterial therapies for stroke.
As with TBI, the formation ROS is a significant source of cytotoxic injury in stroke as well [176]. Although triggered by ischemia, the buildup of ROS and reactive nitrogen species is facilitated by subsequent reperfusion, where ongoing oxygen supply leads to the production of superoxide anions, hydrogen peroxide, and nitric oxide [177]. These free radicals subsequently lead to a cascade of deleterious processes, including BBB breakdown, vasogenic edema, and, eventually, cell death. Measures to counteract the production of these oxidative species has been explored in many early clinical stroke trials, and as in TBI trials these were not effective despite promising preclinical data [178, 179]. Edaravone is another free radical scavenger that has been approved in Japan in the treatment of ischemic strokes and postulated to reduce oxidative damage to cellular membranes, thereby suppressing edema and extension of infarction [180]. A meta-analysis of three trials involving 496 patients suggested a possible signal towards marked neurological improvement, but was limited small sample size [181].
Cortical spreading depression is also an important mediator of secondary neural injury following stroke, and associated with elevations in extracellular potassium and glutamate, which precipitate metabolic crises and ultimately neuronal injury [182]. In ischemic stroke models, studies have shown spreading depolarizations in the penumbra—i.e., peri-infarct depolarizations (PIDs)—as playing a key role in the eventual transition to infarcted core [183]. Transcranial direct current stimulation (tDCS) is now being explored as a therapeutic technology, aimed at utilizing electrical currents to target ischemic neurons and inhibit spreading depolarizations. In animal models of MCAO, cathodal tDCS has been shown to reduce neuronal excitability and metabolic demand in ischemic tissue, thereby reducing infarct volumes by 30–37% [184, 185]. A phase II clinical trial in Europe is now underway, with cathodal tDCS being applied to patients for a duration of 6 h after mechanical embolectomy [186].
Limiting Excitotoxicity
Pharmacological approaches to neuroprotection have in general addressed very specific pathways and time points along the ischemic cascade [104]. Among the more promising of newer single pathway agents are those targeting excitotoxic mechanisms. Excitotoxic neuronal injury is directly related to a surge in extracellular glutamate in ischemic cells. The accumulation of glutamate stimulates a downstream reaction involving the influx of calcium ions and activation of NMDA receptors, as well as other downstream targets (PTEN, DAPK1), ultimately resulting in cell death [187]. Reduced excitotoxicity is a major mechanism of resilience seen in ischemic preconditioning and animal models with innate ischemic tolerance. Strategies to inhibit glutamate-mediated neurotoxicity were the focal point of many earlier clinical trials that failed to show benefit [188, 189] or demonstrated worse outcome [190, 191]. Newer more specific pathways to disrupt glutamate excitotoxicity are being actively investigated, such as NA-1, which limits excitotoxicity by inhibiting the interaction between NMDA receptors and the PSD95 scaffolding protein. NA-1 efficacy is being explored in 2 clinical trials, one in the prehospital setting and a second in the context of endovascular thrombectomy where therapy can be delivered rapidly and locally with minimal side effects [159].
Immune Therapies to Reduce Post-stroke Inflammation
Reduced stroke-related inflammation is key mechanism underlying preconditioning as well as improved outcomes of younger stroke patients [11, 78]. Preconditioning to ischemia can be induced by immune mediators such as TLR4 and TNFα, further reinforcing the importance of these pathways in neuroresilience [45, 192]. The inflammatory response following stroke is an important contributor to acute and even chronic clinical pathology [193, 194]. Ischemic injury triggers an inflammatory cascade beginning with activation of microglia by damage-associated molecules, which mobilizes peripheral immune cells including neutrophils, monocytes, and lymphocytes into the injury site (see Fig. 3). Numerous regulators of macrophage and lymphocytic function can influence the duration and intensity of the immune response [195], and despite a poor understanding of the specifics of these interactions several early phase clinical trials have been performed on drugs with indications for other diseases including minocycline, interleukin-1 receptor antagonist (IL-1ra), fingolimod, and natalizumab. Fingolimod, which limits the infiltration of lymphocytes to the brain, has shown efficacy in two small single-center open-label pilot studies [196, 197]. Another multiple sclerosis therapeutic preventing immune cell entry into the brain, natalizumab, demonstrated mixed results in a phase II trial [198]. It did not achieve its primary end point of reducing infarct growth on MRI at day 5 compared to baseline; however, patients receiving natalizumab did have significantly improved outcomes at 30 days (although not significantly so at 90 days) suggesting that further trials may be helpful in clarifying its effect. Interleukin-1ra, which is used to treat autoimmune conditions such as rheumatoid arthritis, was also investigated in a phase II study [199]. The study achieved its end point of reducing serum concentrations of inflammatory markers at day 3; however, the clinical significance of this is unclear as IL-1ra treatment was not associated with a change in clinical outcome. Minocycline has received a great deal of attention in the preclinical literature as a potent neuroprotectant with multiple beneficial mechanisms of action including reducing apoptosis, MMP expression, and risk of hemorrhage with tPA [200]. In two early phase II trials, it has not shown efficacy, although an additional trial examining minocycline’s ability to reduce hemorrhagic transformation after tPA is still recruiting patients [201,202,203].
Remote ischemic conditioning (RIC) has undergone several clinical trials in myocardial infarction and stroke with thus far mixed results. RIC can be relatively easily implemented using blood pressure cuffs to create brief limb ischemia. It is based on the premise that transient non-lethal ischemia of muscle can protect the brain (or heart) from recent ischemic injury, and may work by modulating inflammatory responses at the site of the injury. In small patient pilot studies, 20 min of forearm ischemia resulted in an increase in CD11b neutrophil expression in circulating blood, while downregulating TLR and TNFα signaling genes as well as leukocyte adhesion and chemotaxis genes [204, 205]. There have been mixed results with regard to RIC in both the myocardial infarction and stroke clinical literature and larger studies with carefully selected patients and optimized RIC regimens are needed. In the largest phase II trial of RIC in stroke (26 patients), encouraging results were demonstrated, as 4 cycles of RIC resulted in an improved NIH stroke scale score at 90 days and significantly increased plasma levels of heat-shock protein-27 [206].
Given the heterogeneity in brain injuries and immune response, treatment of stroke and other brain injuries with immune-modulating agents will require further refinement in our understanding of detrimental inflammatory responses and beneficial recovery-supporting immune responses. Agents may be more effective when patients are selected for treatment at specific times when patient-specific markers suggest immune responses require modulation, rather than treating all patients similarly. Given the often subacute and chronic changes in immune responses observed after brain injury, additional research is necessary to identify the optimal windows of opportunity to provide pharmacologic treatments targeting recovery and autoimmune phenomena as well [194].
Summary and Future Research Directions
Neuroprotection has been defined as a therapeutic strategy to antagonize or slow the biochemical and molecular events leading to irreversible ischemic injury [9]. Over the last decade, our understanding of the scope and duration of the molecular, biochemical, and physiological events surrounding brain injuries has broadened dramatically. We have gained valuable insights into not only how brain injuries affect neurons and glia locally, but also how injury modifies neuronal circuits and immune responses to chronically alter patients’ behavior, cognitive functions, and even mood. As such, the concept of neuroprotection can be strengthened by drawing upon examples of neuroresilience from both the bench and the bedside. Models of innate or induced ischemia tolerance and clinical variability in brain injury recovery point to coordinated role of metabolic, inflammatory, and regenerative pathways in optimizing functional outcomes. It is clear that simply countering the injury cascade over time is not adequate, and therapies broadly anticipating the necessary adaptations and encouraging resilience should be the new standard. Achieving clinically measurable resilience after brain injuries will require concurrent or multifunctional therapeutic approaches acting on multiple pathways delivered at optimal times to encourage persistent brain cell recovery and restore the integrity of neural circuits [93, 207, 208].
Despite massive scientific efforts in developing a neuroprotective agent or approach for TBI or stroke, clinical trials have not yet yielded promising results. Therapeutic agents have to tread a fine line between broadly affecting multiple resilience pathways and risking detrimental systemic side effects, or specifically modulating one pathway and compromising clinical efficacy. The resilience framework is particularly helpful from a translational perspective as it highlights the need for further understanding the interaction between individualized patient responses to injuries and treatments in order to optimize functional outcomes. Identifying these therapeutic opportunities (i.e., a metabolic crisis or aberrant immune response) for intervention in real time is a major goal of multimodality neuromonitoring [89]. Over the next decade, we are bound to see a new wave of individualized treatments as translational aspects of novel biomarkers, brain imaging, and machine-learning algorithms are coupled with emerging therapeutics such as CRISPR-based gene and RNA editing technologies and autologous stem cell delivery [209]. Additional technology development and clinical trials in these exciting areas of neurocritical care will guide the next generation of neuroresilience therapies and greatly improve the outcomes of patients suffering from brain injuries.
References
Andrews PJ, Sinclair HL, Rodriguez A, Harris BA, Battison CG, Rhodes JK, et al. Hypothermia for Intracranial Hypertension after Traumatic Brain Injury. N Engl J Med. 2015;373(25):2403-12.
Jovin TG, Albers GW, Liebeskind DS, Consortium SI. Stroke Treatment Academic Industry Roundtable: The Next Generation of Endovascular Trials. Stroke. 2016;47(10):2656-65.
Liu S, Zhen G, Meloni BP, Campbell K, Winn HR. Rodent Stroke Model Guidelines for Preclinical Stroke Trials (1st Edition). J Exp Stroke Transl Med. 2009;2(2):2-27.
Stroke Therapy Academic Industry R. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30(12):2752-8.
Margulies S, Anderson G, Atif F, Badaut J, Clark R, Empey P, et al. Combination Therapies for Traumatic Brain Injury: Retrospective Considerations. J Neurotrauma. 2016;33(1):101-12.
Stoica B, Byrnes K, Faden AI. Multifunctional drug treatment in neurotrauma. Neurotherapeutics. 2009;6(1):14-27.
Vink R, Nimmo AJ. Multifunctional drugs for head injury. Neurotherapeutics. 2009;6(1):28-42.
Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ. Neurobiology of resilience. Nat Neurosci. 2012;15(11):1475-84.
Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55(3):363-89.
Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7(6):437-48.
Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat Neurosci. 2011;14(11):1363-8.
Schurr A, Reid KH, Tseng MT, West C, Rigor BM. Adaptation of adult brain tissue to anoxia and hypoxia in vitro. Brain Res. 1986;374(2):244-8.
Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, et al. 'Ischemic tolerance' phenomenon found in the brain. Brain Res. 1990;528(1):21-4.
Nishio S, Chen ZF, Yunoki M, Toyoda T, Anzivino M, Lee KS. Hypothermia-induced ischemic tolerance. Ann N Y Acad Sci. 1999;890:26-41.
Plamondon H, Blondeau N, Heurteaux C, Lazdunski M. Mutually protective actions of kainic acid epileptic preconditioning and sublethal global ischemia on hippocampal neuronal death: involvement of adenosine A1 receptors and K(ATP) channels. J Cereb Blood Flow Metab. 1999;19(12):1296-308.
Anrather J, Hallenbeck JM. Biological networks in ischemic tolerance - rethinking the approach to clinical conditioning. Transl Stroke Res. 2013;4(1):114-29.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111-22.
Marti HH. Erythropoietin and the hypoxic brain. J Exp Biol. 2004;207(Pt 18):3233-42.
Lee ST, Chu K, Jung KH, Yoon HJ, Jeon D, Kang KM, et al. MicroRNAs induced during ischemic preconditioning. Stroke. 2010;41(8):1646-51.
Park HK, Chu K, Jung KH, Lee ST, Bahn JJ, Kim M, et al. Autophagy is involved in the ischemic preconditioning. Neurosci Lett. 2009;451(1):16-9.
Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, et al. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2012;8(3):310-25.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892-903.
Acker T, Acker H. Cellular oxygen sensing need in CNS function: physiological and pathological implications. J Exp Biol. 2004;207(Pt 18):3171-88.
Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci. 2003;23(2):384-91.
Raval AP, Dave KR, DeFazio RA, Perez-Pinzon MA. epsilonPKC phosphorylates the mitochondrial K(+) (ATP) channel during induction of ischemic preconditioning in the rat hippocampus. Brain Res. 2007;1184:345-53.
Truettner J, Busto R, Zhao W, Ginsberg MD, Perez-Pinzon MA. Effect of ischemic preconditioning on the expression of putative neuroprotective genes in the rat brain. Brain Res Mol Brain Res. 2002;103(1-2):106-15.
Yanamoto H, Mizuta I, Nagata I, Xue J, Zhang Z, Kikuchi H. Infarct tolerance accompanied enhanced BDNF-like immunoreactivity in neuronal nuclei. Brain Res. 2000;877(2):331-44.
Corbett D, Giles T, Evans S, McLean J, Biernaskie J. Dynamic changes in CA1 dendritic spines associated with ischemic tolerance. Exp Neurol. 2006;202(1):133-8.
Neumann JT, Thompson JW, Raval AP, Cohan CH, Koronowski KB, Perez-Pinzon MA. Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection. J Cereb Blood Flow Metab. 2015;35(1):121-30.
Thompson JW, Dave KR, Young JI, Perez-Pinzon MA. Ischemic preconditioning alters the epigenetic profile of the brain from ischemic intolerance to ischemic tolerance. Neurotherapeutics. 2013;10(4):789-97.
Cuomo O, Pignataro G, Sirabella R, Molinaro P, Anzilotti S, Scorziello A, et al. Sumoylation of LYS590 of NCX3 f-Loop by SUMO1 Participates in Brain Neuroprotection Induced by Ischemic Preconditioning. Stroke. 2016;47(4):1085-93.
Lee YJ, Castri P, Bembry J, Maric D, Auh S, Hallenbeck JM. SUMOylation participates in induction of ischemic tolerance. J Neurochem. 2009;109(1):257-67.
Yang W, Sheng H, Warner DS, Paschen W. Transient global cerebral ischemia induces a massive increase in protein sumoylation. J Cereb Blood Flow Metab. 2008;28(2):269-79.
Zhang H, Huang D, Zhou J, Yue Y, Wang X. SUMOylation participates in induction of ischemic tolerance in mice. Brain Res Bull. 2019;147:159-64.
Zhang L, Liu X, Sheng H, Liu S, Li Y, Zhao JQ, et al. Neuron-specific SUMO knockdown suppresses global gene expression response and worsens functional outcome after transient forebrain ischemia in mice. Neuroscience. 2017;343:190-212.
Cimarosti H, Lindberg C, Bomholt SF, Ronn LC, Henley JM. Increased protein SUMOylation following focal cerebral ischemia. Neuropharmacology. 2008;54(2):280-9.
Datwyler AL, Lattig-Tunnemann G, Yang W, Paschen W, Lee SL, Dirnagl U, et al. SUMO2/3 conjugation is an endogenous neuroprotective mechanism. J Cereb Blood Flow Metab. 2011;31(11):2152-9.
Silveirinha V, Stephens GJ, Cimarosti H. Molecular targets underlying SUMO-mediated neuroprotection in brain ischemia. J Neurochem. 2013;127(5):580-91.
Lee YJ, Bernstock JD, Nagaraja N, Ko B, Hallenbeck JM. Global SUMOylation facilitates the multimodal neuroprotection afforded by quercetin against the deleterious effects of oxygen/glucose deprivation and the restoration of oxygen/glucose. J Neurochem. 2016;138(1):101-16.
Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14(4):340-5.
Shalizi A, Gaudilliere B, Yuan Z, Stegmuller J, Shirogane T, Ge Q, et al. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311(5763):1012-7.
Tatham MH, Matic I, Mann M, Hay RT. Comparative proteomic analysis identifies a role for SUMO in protein quality control. Sci Signal. 2011;4(178):rs4.
Anrather J, Iadecola C. Inflammation and Stroke: An Overview. Neurotherapeutics. 2016;13(4):661-70.
Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL, et al. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199(1-2):75-82.
Pradillo JM, Fernandez-Lopez D, Garcia-Yebenes I, Sobrado M, Hurtado O, Moro MA, et al. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J Neurochem. 2009;109(1):287-94.
Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, et al. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci. 2011;31(23):8456-63.
Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26(6):1880-7.
Norden DM, Fenn AM, Dugan A, Godbout JP. TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia. 2014;62(6):881-95.
Shinozaki Y, Shibata K, Yoshida K, Shigetomi E, Gachet C, Ikenaka K, et al. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia via P2Y1 Receptor Downregulation. Cell Rep. 2017;19(6):1151-64.
Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, et al. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke. 2004;35(3):616-21.
Frerichs KU, Kennedy C, Sokoloff L, Hallenbeck JM. Local cerebral blood flow during hibernation, a model of natural tolerance to "cerebral ischemia". J Cereb Blood Flow Metab. 1994;14(2):193-205.
Dave KR, Prado R, Raval AP, Drew KL, Perez-Pinzon MA. The arctic ground squirrel brain is resistant to injury from cardiac arrest during euthermia. Stroke. 2006;37(5):1261-5.
Bhowmick S, Moore JT, Kirschner DL, Drew KL. Arctic ground squirrel hippocampus tolerates oxygen glucose deprivation independent of hibernation season even when not hibernating and after ATP depletion, acidosis, and glutamate efflux. J Neurochem. 2017;142(1):160-70.
Bhowmick S, Drew KL. Arctic ground squirrel resist peroxynitrite-mediated cell death in response to oxygen glucose deprivation. Free Radic Biol Med. 2017;113:203-11.
von der Ohe CG, Darian-Smith C, Garner CC, Heller HC. Ubiquitous and temperature-dependent neural plasticity in hibernators. J Neurosci. 2006;26(41):10590-8.
Ou J, Ball JM, Luan Y, Zhao T, Miyagishima KJ, Xu Y, et al. iPSCs from a Hibernator Provide a Platform for Studying Cold Adaptation and Its Potential Medical Applications. Cell. 2018;173(4):851-63 e16.
Guo Q, Mi X, Sun X, Li X, Fu W, Xu S, et al. Remarkable plasticity of Na(+), K(+)-ATPase, Ca(2+)-ATPase and SERCA contributes to muscle disuse atrophy resistance in hibernating Daurian ground squirrels. Sci Rep. 2017;7(1):10509.
Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003;362(9389):1028-37.
Drew KL, Harris MB, LaManna JC, Smith MA, Zhu XW, Ma YL. Hypoxia tolerance in mammalian heterotherms. J Exp Biol. 2004;207(Pt 18):3155-62.
Drew KL, Buck CL, Barnes BM, Christian SL, Rasley BT, Harris MB. Central nervous system regulation of mammalian hibernation: implications for metabolic suppression and ischemia tolerance. J Neurochem. 2007;102(6):1713-26.
Christian SL, Ross AP, Zhao HW, Kristenson HJ, Zhan X, Rasley BT, et al. Arctic ground squirrel (Spermophilus parryii) hippocampal neurons tolerate prolonged oxygen-glucose deprivation and maintain baseline ERK1/2 and JNK activation despite drastic ATP loss. J Cereb Blood Flow Metab. 2008;28(7):1307-19.
Dave KR, Anthony Defazio R, Raval AP, Dashkin O, Saul I, Iceman KE, et al. Protein kinase C epsilon activation delays neuronal depolarization during cardiac arrest in the euthermic arctic ground squirrel. J Neurochem. 2009;110(4):1170-9.
Lee YJ, Miyake S, Wakita H, McMullen DC, Azuma Y, Auh S, et al. Protein SUMOylation is massively increased in hibernation torpor and is critical for the cytoprotection provided by ischemic preconditioning and hypothermia in SHSY5Y cells. J Cereb Blood Flow Metab. 2007;27(5):950-62.
Ma YL, Zhu X, Rivera PM, Toien O, Barnes BM, LaManna JC, et al. Absence of cellular stress in brain after hypoxia induced by arousal from hibernation in Arctic ground squirrels. Am J Physiol Regul Integr Comp Physiol. 2005;289(5):R1297-306.
Ni Z, Storey KB. Heme oxygenase expression and Nrf2 signaling during hibernation in ground squirrels. Can J Physiol Pharmacol. 2010;88(3):379-87.
Andrews MT. Molecular interactions underpinning the phenotype of hibernation in mammals. J Exp Biol. 2019;222(Pt 2).
Marquez de la Plata CD, Hart T, Hammond FM, Frol AB, Hudak A, Harper CR, et al. Impact of age on long-term recovery from traumatic brain injury. Arch Phys Med Rehabil. 2008;89(5):896-903.
Duhaime AC, Hunter JV, Grate LL, Kim A, Golden J, Demidenko E, et al. Magnetic resonance imaging studies of age-dependent responses to scaled focal brain injury in the piglet. J Neurosurg. 2003;99(3):542-8.
Missios S, Harris BT, Dodge CP, Simoni MK, Costine BA, Lee YL, et al. Scaled cortical impact in immature swine: effect of age and gender on lesion volume. J Neurotrauma. 2009;26(11):1943-51.
Brickler TR, Hazy A, Guilhaume Correa F, Dai R, Kowalski EJA, Dickerson R, et al. Angiopoietin/Tie2 Axis Regulates the Age-at-Injury Cerebrovascular Response to Traumatic Brain Injury. J Neurosci. 2018;38(45):9618-34.
Claus CP, Tsuru-Aoyagi K, Adwanikar H, Walker B, Manvelyan H, Whetstone W, et al. Age is a determinant of leukocyte infiltration and loss of cortical volume after traumatic brain injury. Dev Neurosci. 2010;32(5-6):454-65.
Fan P, Yamauchi T, Noble LJ, Ferriero DM. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma. 2003;20(5):437-45.
Giza CC, Prins ML. Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev Neurosci. 2006;28(4-5):364-79.
Kamper JE, Pop V, Fukuda AM, Ajao DO, Hartman RE, Badaut J. Juvenile traumatic brain injury evolves into a chronic brain disorder: behavioral and histological changes over 6months. Exp Neurol. 2013;250:8-19.
Moretti R, Pansiot J, Bettati D, Strazielle N, Ghersi-Egea JF, Damante G, et al. Blood-brain barrier dysfunction in disorders of the developing brain. Front Neurosci. 2015;9:40.
Pop V, Sorensen DW, Kamper JE, Ajao DO, Murphy MP, Head E, et al. Early brain injury alters the blood-brain barrier phenotype in parallel with beta-amyloid and cognitive changes in adulthood. J Cereb Blood Flow Metab. 2013;33(2):205-14.
Moraga A, Pradillo JM, Garcia-Culebras A, Palma-Tortosa S, Ballesteros I, Hernandez-Jimenez M, et al. Aging increases microglial proliferation, delays cell migration, and decreases cortical neurogenesis after focal cerebral ischemia. J Neuroinflammation. 2015;12:87.
Ritzel RM, Lai YJ, Crapser JD, Patel AR, Schrecengost A, Grenier JM, et al. Aging alters the immunological response to ischemic stroke. Acta Neuropathol. 2018;136(1):89-110.
Soderholm M, Pedersen A, Lorentzen E, Stanne TM, Bevan S, Olsson M, et al. Genome-wide association meta-analysis of functional outcome after ischemic stroke. Neurology. 2019;92(12):e1271-e83.
Garnier P, Ying W, Swanson RA. Ischemic preconditioning by caspase cleavage of poly(ADP-ribose) polymerase-1. J Neurosci. 2003;23(22):7967-73.
Kauppinen TM, Swanson RA. The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience. 2007;145(4):1267-72.
Zhang Y, Zhang X, Park TS, Gidday JM. Cerebral endothelial cell apoptosis after ischemia-reperfusion: role of PARP activation and AIF translocation. J Cereb Blood Flow Metab. 2005;25(7):868-77.
Zeiler FA, McFadyen C, Newcombe VFJ, Synnot A, Donoghue EL, Ripatti S, et al. Genetic Influences on Patient-Oriented Outcomes in Traumatic Brain Injury: A Living Systematic Review of Non-Apolipoprotein E Single-Nucleotide Polymorphisms. J Neurotrauma. 2018;35:1–17.
Zeiler FA, Thelin EP, Donnelly J, Stevens AR, Smielewski P, Czosnyka M, et al. Genetic drivers of cerebral blood flow dysfunction in TBI: a speculative synthesis. Nat Rev Neurol. 2019;15(1):25-39.
Centers for Disease C, Prevention. CDC grand rounds: reducing severe traumatic brain injury in the United States. MMWR Morb Mortal Wkly Rep. 2013;62(27):549-52.
Gardner RC, Dams-O'Connor K, Morrissey MR, Manley GT. Geriatric Traumatic Brain Injury: Epidemiology, Outcomes, Knowledge Gaps, and Future Directions. J Neurotrauma. 2018;35(7):889–906.
Peeters W, van den Brande R, Polinder S, Brazinova A, Steyerberg EW, Lingsma HF, et al. Epidemiology of traumatic brain injury in Europe. Acta Neurochir (Wien). 2015;157(10):1683-96.
Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015;72(3):355-62.
Le Roux P, Menon DK, Citerio G, Vespa P, Bader MK, Brophy GM, et al. Consensus summary statement of the International Multidisciplinary Consensus Conference on Multimodality Monitoring in Neurocritical Care: a statement for healthcare professionals from the Neurocritical Care Society and the European Society of Intensive Care Medicine. Neurocrit Care. 2014;21 Suppl 2:S1-26.
Hu X, Xu P, Asgari S, Vespa P, Bergsneider M. Forecasting ICP elevation based on prescient changes of intracranial pressure waveform morphology. IEEE Trans Biomed Eng. 2010;57(5):1070-8.
Lazaridis C, Rusin CG, Robertson CS. Secondary brain injury: Predicting and preventing insults. Neuropharmacology. 2019;145(Pt B):145-52.
Kramer AH, Couillard PL, Zygun DA, Aries MJ, Gallagher CN. Continuous Assessment of "Optimal" Cerebral Perfusion Pressure in Traumatic Brain Injury: A Cohort Study of Feasibility, Reliability, and Relation to Outcome. Neurocrit Care. 2019;30(1):51-61.
Zoerle T, Carbonara M, Zanier ER, Ortolano F, Bertani G, Magnoni S, et al. Rethinking Neuroprotection in Severe Traumatic Brain Injury: Toward Bedside Neuroprotection. Front Neurol. 2017;8:354.
Deutsch ER, Espinoza TR, Atif F, Woodall E, Kaylor J, Wright DW. Progesterone's role in neuroprotection, a review of the evidence. Brain Res. 2013;1530:82-105.
Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD, et al. A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med. 2014;371(26):2467-76.
Wright DW, Yeatts SD, Silbergleit R, Palesch YY, Hertzberg VS, Frankel M, et al. Very early administration of progesterone for acute traumatic brain injury. N Engl J Med. 2014;371(26):2457-66.
Cooper DJ, Nichol AD, Bailey M, Bernard S, Cameron PA, Pili-Floury S, et al. Effect of Early Sustained Prophylactic Hypothermia on Neurologic Outcomes Among Patients With Severe Traumatic Brain Injury: The POLAR Randomized Clinical Trial. JAMA. 2018;320(21):2211-20.
Maekawa T, Yamashita S, Nagao S, Hayashi N, Ohashi Y, Brain-Hypothermia Study G. Prolonged mild therapeutic hypothermia versus fever control with tight hemodynamic monitoring and slow rewarming in patients with severe traumatic brain injury: a randomized controlled trial. J Neurotrauma. 2015;32(7):422-9.
Menon DK, Maas AI. Traumatic brain injury in 2014. Progress, failures and new approaches for TBI research. Nat Rev Neurol. 2015;11(2):71-2.
Stein DG. Embracing failure: What the Phase III progesterone studies can teach about TBI clinical trials. Brain Inj. 2015;29(11):1259-72.
Andrews PJ, Sinclair HL, Rodriguez A, Harris B, Rhodes J, Watson H, et al. Therapeutic hypothermia to reduce intracranial pressure after traumatic brain injury: the Eurotherm3235 RCT. Health Technol Assess. 2018;22(45):1-134.
McKee CA, Lukens JR. Emerging Roles for the Immune System in Traumatic Brain Injury. Front Immunol. 2016;7:556.
Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13(3):171-91.
Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391-7.
Vespa P, Tubi M, Claassen J, Buitrago-Blanco M, McArthur D, Velazquez AG, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol. 2016;79(4):579-90.
Kilbaugh TJ, Karlsson M, Byro M, Bebee A, Ralston J, Sullivan S, et al. Mitochondrial bioenergetic alterations after focal traumatic brain injury in the immature brain. Exp Neurol. 2015;271:136-44.
Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. 2012;15(10):1407-13.
Walsh JJ, Huang Y, Simmons JW, Goodrich JA, McHugh B, Rothman DL, et al. Dynamic Thermal Mapping of Localized Therapeutic Hypothermia in the Brain. J Neurotrauma. 2020;37(1):55–65.
Yenari MA, Han HS. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat Rev Neurosci. 2012;13(4):267-78.
Pandya JD, Leung LY, Yang X, Flerlage WJ, Gilsdorf JS, Deng-Bryant Y, et al. Comprehensive Profile of Acute Mitochondrial Dysfunction in a Preclinical Model of Severe Penetrating TBI. Front Neurol. 2019;10:605.
Jalloh I, Helmy A, Howe DJ, Shannon RJ, Grice P, Mason A, et al. Focally perfused succinate potentiates brain metabolism in head injury patients. J Cereb Blood Flow Metab. 2017;37(7):2626-38.
Prins ML, Matsumoto JH. The collective therapeutic potential of cerebral ketone metabolism in traumatic brain injury. J Lipid Res. 2014;55(12):2450-7.
Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. 2008;86(8):1812-22.
Mahmoodpoor A, Shokouhi G, Hamishehkar H, Soleimanpour H, Sanaie S, Porhomayon J, et al. A pilot trial of l-carnitine in patients with traumatic brain injury: Effects on biomarkers of injury. J Crit Care. 2018;45:128-32.
Marshall LF, Maas AI, Marshall SB, Bricolo A, Fearnside M, Iannotti F, et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg. 1998;89(4):519-25.
Muizelaar JP, Kupiec JW, Rapp LA. PEG-SOD after head injury. J Neurosurg. 1995;83(5):942.
Chandran R, Kim T, Mehta SL, Udho E, Chanana V, Cengiz P, et al. A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J Cereb Blood Flow Metab. 2018;38(10):1818-27.
Chao H, Lin C, Zuo Q, Liu Y, Xiao M, Xu X, et al. Cardiolipin-Dependent Mitophagy Guides Outcome after Traumatic Brain Injury. J Neurosci. 2019;39(10):1930-43.
Ho KM, Honeybul S, Yip CB, Silbert BI. Prognostic significance of blood-brain barrier disruption in patients with severe nonpenetrating traumatic brain injury requiring decompressive craniectomy. J Neurosurg. 2014;121(3):674-9.
Baskaya MK, Rao AM, Dogan A, Donaldson D, Dempsey RJ. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett. 1997;226(1):33-6.
Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6(7):393-403.
Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2(4):492-516.
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409-27.
Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017;20(3):406-16.
Cheng J, Korte N, Nortley R, Sethi H, Tang Y, Attwell D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018;136(4):507-23.
Erdo F, Denes L, de Lange E. Age-associated physiological and pathological changes at the blood-brain barrier: A review. J Cereb Blood Flow Metab. 2017;37(1):4-24.
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11(1):56-64.
Gennai S, Monsel A, Hao Q, Liu J, Gudapati V, Barbier EL, et al. Cell-based therapy for traumatic brain injury. Br J Anaesth. 2015;115(2):203-12.
Tajiri N, Kaneko Y, Shinozuka K, Ishikawa H, Yankee E, McGrogan M, et al. Stem cell recruitment of newly formed host cells via a successful seduction? Filling the gap between neurogenic niche and injured brain site. PLoS One. 2013;8(9):e74857.
Galindo LT, Filippo TR, Semedo P, Ariza CB, Moreira CM, Camara NO, et al. Mesenchymal stem cell therapy modulates the inflammatory response in experimental traumatic brain injury. Neurol Res Int. 2011;2011:564089.
Li Y, Yang YY, Ren JL, Xu F, Chen FM, Li A. Exosomes secreted by stem cells from human exfoliated deciduous teeth contribute to functional recovery after traumatic brain injury by shifting microglia M1/M2 polarization in rats. Stem Cell Res Ther. 2017;8(1):198.
Ni H, Yang S, Siaw-Debrah F, Hu J, Wu K, He Z, et al. Exosomes Derived From Bone Mesenchymal Stem Cells Ameliorate Early Inflammatory Responses Following Traumatic Brain Injury. Front Neurosci. 2019;13:14.
Patel NA, Moss LD, Lee JY, Tajiri N, Acosta S, Hudson C, et al. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J Neuroinflammation. 2018;15(1):204.
Kondziella D, Fisher PM, Larsen VA, Hauerberg J, Fabricius M, Moller K, et al. Functional MRI for Assessment of the Default Mode Network in Acute Brain Injury. Neurocrit Care. 2017;27(3):401-6.
Palacios EM, Yuh EL, Chang YS, Yue JK, Schnyer DM, Okonkwo DO, et al. Resting-State Functional Connectivity Alterations Associated with Six-Month Outcomes in Mild Traumatic Brain Injury. J Neurotrauma. 2017;34(8):1546-57.
Sharp DJ, Beckmann CF, Greenwood R, Kinnunen KM, Bonnelle V, De Boissezon X, et al. Default mode network functional and structural connectivity after traumatic brain injury. Brain. 2011;134(Pt 8):2233-47.
Shumskaya E, Andriessen TM, Norris DG, Vos PE. Abnormal whole-brain functional networks in homogeneous acute mild traumatic brain injury. Neurology. 2012;79(2):175-82.
Ahmadzadeh H, Smith DH, Shenoy VB. Mechanical Effects of Dynamic Binding between Tau Proteins on Microtubules during Axonal Injury. Biophys J. 2015;109(11):2328-37.
Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, et al. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23(18):6972-81.
Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. 2017;8(1):1000.
Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ. Potential of the Antibody Against cis-Phosphorylated Tau in the Early Diagnosis, Treatment, and Prevention of Alzheimer Disease and Brain Injury. JAMA Neurol. 2016;73(11):1356-62.
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135(10):e146-e603.
Saver JL, Goyal M, van der Lugt A, Menon BK, Majoie CB, Dippel DW, et al. Time to Treatment With Endovascular Thrombectomy and Outcomes From Ischemic Stroke: A Meta-analysis. JAMA. 2016;316(12):1279-88.
Campbell BCV, Ma H, Ringleb PA, Parsons MW, Churilov L, Bendszus M, et al. Extending thrombolysis to 4.5-9 h and wake-up stroke using perfusion imaging: a systematic review and meta-analysis of individual patient data. Lancet. 2019;394(10193):139-47.
Hacke W, Donnan G, Fieschi C, Kaste M, von Kummer R, Broderick JP, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363(9411):768-74.
Ma H, Campbell BCV, Churilov L. Thrombolysis up to 9 Hours after Onset of Stroke. Reply. N Engl J Med. 2019;381(5):488-9.
Seners P, Turc G, Maier B, Mas JL, Oppenheim C, Baron JC. Incidence and Predictors of Early Recanalization After Intravenous Thrombolysis: A Systematic Review and Meta-Analysis. Stroke. 2016;47(9):2409-12.
Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372(1):11-20.
Campbell BC, Mitchell PJ, Kleinig TJ, Dewey HM, Churilov L, Yassi N, et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med. 2015;372(11):1009-18.
Goyal M, Demchuk AM, Menon BK, Eesa M, Rempel JL, Thornton J, et al. Randomized assessment of rapid endovascular treatment of ischemic stroke. N Engl J Med. 2015;372(11):1019-30.
Jovin TG, Chamorro A, Cobo E, de Miquel MA, Molina CA, Rovira A, et al. Thrombectomy within 8 hours after symptom onset in ischemic stroke. N Engl J Med. 2015;372(24):2296-306.
Saver JL, Goyal M, Bonafe A, Diener HC, Levy EI, Pereira VM, et al. Solitaire with the Intention for Thrombectomy as Primary Endovascular Treatment for Acute Ischemic Stroke (SWIFT PRIME) trial: protocol for a randomized, controlled, multicenter study comparing the Solitaire revascularization device with IV tPA with IV tPA alone in acute ischemic stroke. Int J Stroke. 2015;10(3):439-48.
Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet. 2016;387(10029):1723-31.
Albers GW, Marks MP, Kemp S, Christensen S, Tsai JP, Ortega-Gutierrez S, et al. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. N Engl J Med. 2018;378(8):708-18.
Nogueira RG, Jadhav AP, Haussen DC, Bonafe A, Budzik RF, Bhuva P, et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N Engl J Med. 2018;378(1):11-21.
Liebeskind DS, Kasner SE. Neuroprotection for ischaemic stroke: an unattainable goal? CNS Drugs. 2001;15(3):165-74.
Savitz SI, Baron JC, Yenari MA, Sanossian N, Fisher M. Reconsidering Neuroprotection in the Reperfusion Era. Stroke. 2017;48(12):3413-9.
Liebeskind DS, Derdeyn CP, Wechsler LR, Consortium SX. STAIR X: Emerging Considerations in Developing and Evaluating New Stroke Therapies. Stroke. 2018;49(9):2241-7.
Tymianski M. Combining Neuroprotection With Endovascular Treatment of Acute Stroke: Is There Hope? Stroke. 2017;48(6):1700-5.
McBride DW, Zhang JH. Precision Stroke Animal Models: the Permanent MCAO Model Should Be the Primary Model, Not Transient MCAO. Transl Stroke Res. 2017;8(5):397–404.
Shi L, Rocha M, Leak RK, Zhao J, Bhatia TN, Mu H, et al. A new era for stroke therapy: Integrating neurovascular protection with optimal reperfusion. J Cereb Blood Flow Metab. 2018;38(12):2073-91.
Saver JL, Starkman S, Eckstein M, Stratton SJ, Pratt FD, Hamilton S, et al. Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N Engl J Med. 2015;372(6):528-36.
O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59(3):467-77.
McManus M, Liebeskind DS. Blood Pressure in Acute Ischemic Stroke. J Clin Neurol. 2016;12(2):137-46.
Ginsberg MD, Palesch YY, Hill MD, Martin RH, Moy CS, Barsan WG, et al. High-dose albumin treatment for acute ischaemic stroke (ALIAS) Part 2: a randomised, double-blind, phase 3, placebo-controlled trial. Lancet Neurol. 2013;12(11):1049-58.
Campbell BCV, Majoie C, Albers GW, Menon BK, Yassi N, Sharma G, et al. Penumbral imaging and functional outcome in patients with anterior circulation ischaemic stroke treated with endovascular thrombectomy versus medical therapy: a meta-analysis of individual patient-level data. Lancet Neurol. 2019;18(1):46-55.
Hamann GF, Burggraf D, Martens HK, Liebetrau M, Jager G, Wunderlich N, et al. Mild to moderate hypothermia prevents microvascular basal lamina antigen loss in experimental focal cerebral ischemia. Stroke. 2004;35(3):764-9.
Nagel S, Su Y, Horstmann S, Heiland S, Gardner H, Koziol J, et al. Minocycline and hypothermia for reperfusion injury after focal cerebral ischemia in the rat: effects on BBB breakdown and MMP expression in the acute and subacute phase. Brain Res. 2008;1188:198-206.
van der Worp HB, Sena ES, Donnan GA, Howells DW, Macleod MR. Hypothermia in animal models of acute ischaemic stroke: a systematic review and meta-analysis. Brain. 2007;130(Pt 12):3063-74.
De Georgia MA, Krieger DW, Abou-Chebl A, Devlin TG, Jauss M, Davis SM, et al. Cooling for Acute Ischemic Brain Damage (COOL AID): a feasibility trial of endovascular cooling. Neurology. 2004;63(2):312-7.
Hemmen TM, Raman R, Guluma KZ, Meyer BC, Gomes JA, Cruz-Flores S, et al. Intravenous thrombolysis plus hypothermia for acute treatment of ischemic stroke (ICTuS-L): final results. Stroke. 2010;41(10):2265-70.
Lyden P, Hemmen T, Grotta J, Rapp K, Ernstrom K, Rzesiewicz T, et al. Results of the ICTuS 2 Trial (Intravascular Cooling in the Treatment of Stroke 2). Stroke. 2016;47(12):2888-95.
Horn CM, Sun CH, Nogueira RG, Patel VN, Krishnan A, Glenn BA, et al. Endovascular Reperfusion and Cooling in Cerebral Acute Ischemia (ReCCLAIM I). J Neurointerv Surg. 2014;6(2):91-5.
Hong JM, Lee JS, Song HJ, Jeong HS, Choi HA, Lee K. Therapeutic hypothermia after recanalization in patients with acute ischemic stroke. Stroke. 2014;45(1):134-40.
Chen J, Liu L, Zhang H, Geng X, Jiao L, Li G, et al. Endovascular Hypothermia in Acute Ischemic Stroke: Pilot Study of Selective Intra-Arterial Cold Saline Infusion. Stroke. 2016;47(7):1933-5.
Watanabe T, Tahara M, Todo S. The novel antioxidant edaravone: from bench to bedside. Cardiovasc Ther. 2008;26(2):101-14.
Chamorro A, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016;15(8):869-81.
Lees KR, Zivin JA, Ashwood T, Davalos A, Davis SM, Diener HC, et al. NXY-059 for acute ischemic stroke. N Engl J Med. 2006;354(6):588-600.
Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, et al. NXY-059 for the treatment of acute ischemic stroke. N Engl J Med. 2007;357(6):562-71.
Edaravone Acute Infarction Study G. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc Dis. 2003;15(3):222-9.
Feng S, Yang Q, Liu M, Li W, Yuan W, Zhang S, et al. Edaravone for acute ischaemic stroke. Cochrane Database Syst Rev. 2011(12):CD007230.
Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17(4):439-47.
Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129(Pt 3):778-90.
Notturno F, Pace M, Zappasodi F, Cam E, Bassetti CL, Uncini A. Neuroprotective effect of cathodal transcranial direct current stimulation in a rat stroke model. J Neurol Sci. 2014;342(1-2):146-51.
Peruzzotti-Jametti L, Cambiaghi M, Bacigaluppi M, Gallizioli M, Gaude E, Mari S, et al. Safety and efficacy of transcranial direct current stimulation in acute experimental ischemic stroke. Stroke. 2013;44(11):3166-74.
Pruvost-Robieux E, Calvet D, Ben Hassen W, Turc G, Marchi A, Mele N, et al. Design and Methodology of a Pilot Randomized Controlled Trial of Transcranial Direct Current Stimulation in Acute Middle Cerebral Artery Stroke (STICA). Front Neurol. 2018;9:816.
Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157-88.
Horn J, de Haan RJ, Vermeulen M, Limburg M. Very Early Nimodipine Use in Stroke (VENUS): a randomized, double-blind, placebo-controlled trial. Stroke. 2001;32(2):461-5.
Wahlgren NG, Ranasinha KW, Rosolacci T, Franke CL, van Erven PM, Ashwood T, et al. Clomethiazole acute stroke study (CLASS): results of a randomized, controlled trial of clomethiazole versus placebo in 1360 acute stroke patients. Stroke. 1999;30(1):21-8.
Albers GW, Goldstein LB, Hall D, Lesko LM, Aptiganel Acute Stroke I. Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA. 2001;286(21):2673-82.
Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, et al. Selfotel in acute ischemic stroke : possible neurotoxic effects of an NMDA antagonist. Stroke. 2000;31(2):347-54.
Watters O, O'Connor JJ. A role for tumor necrosis factor-alpha in ischemia and ischemic preconditioning. J Neuroinflammation. 2011;8:87.
Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8(7):401-10.
Doyle KP, Buckwalter MS. Does B lymphocyte-mediated autoimmunity contribute to post-stroke dementia? Brain Behav Immun. 2017;64:1-8.
Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2009;158(3):1174-83.
Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111(51):18315-20.
Tian DC, Shi K, Zhu Z, Yao J, Yang X, Su L, et al. Fingolimod enhances the efficacy of delayed alteplase administration in acute ischemic stroke by promoting anterograde reperfusion and retrograde collateral flow. Ann Neurol. 2018;84(5):717-28.
Elkins J, Veltkamp R, Montaner J, Johnston SC, Singhal AB, Becker K, et al. Safety and efficacy of natalizumab in patients with acute ischaemic stroke (ACTION): a randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2017;16(3):217-26.
Smith CJ, Hulme S, Vail A, Heal C, Parry-Jones AR, Scarth S, et al. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): A Randomized Controlled Phase 2 Trial. Stroke. 2018;49(5):1210-6.
Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, Lo EH. Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke. 2008;39(12):3372-7.
Blacker DJ, Prentice D, Alvaro A, Bates TR, Bynevelt M, Kelly A, et al. Reducing haemorrhagic transformation after thrombolysis for stroke: a strategy utilising minocycline. Stroke Res Treat. 2013;2013:362961.
Fagan SC, Waller JL, Nichols FT, Edwards DJ, Pettigrew LC, Clark WM, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke. 2010;41(10):2283-7.
Kohler E, Prentice DA, Bates TR, Hankey GJ, Claxton A, van Heerden J, et al. Intravenous minocycline in acute stroke: a randomized, controlled pilot study and meta-analysis. Stroke. 2013;44(9):2493-9.
Kharbanda RK, Peters M, Walton B, Kattenhorn M, Mullen M, Klein N, et al. Ischemic preconditioning prevents endothelial injury and systemic neutrophil activation during ischemia-reperfusion in humans in vivo. Circulation. 2001;103(12):1624-30.
Konstantinov IE, Arab S, Kharbanda RK, Li J, Cheung MM, Cherepanov V, et al. The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol Genomics. 2004;19(1):143-50.
England TJ, Hedstrom A, O'Sullivan S, Donnelly R, Barrett DA, Sarmad S, et al. RECAST (Remote Ischemic Conditioning After Stroke Trial): A Pilot Randomized Placebo Controlled Phase II Trial in Acute Ischemic Stroke. Stroke. 2017;48(5):1412-5.
George PM, Steinberg GK. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron. 2015;87(2):297-309.
Silasi G, Murphy TH. Stroke and the connectome: how connectivity guides therapeutic intervention. Neuron. 2014;83(6):1354-68.
Park H, Oh J, Shim G, Cho B, Chang Y, Kim S, et al. In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer's disease. Nat Neurosci. 2019;22(4):524-8.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Singhal, N.S., Sun, CH., Lee, E.M. et al. Resilience to Injury: A New Approach to Neuroprotection?. Neurotherapeutics 17, 457–474 (2020). https://doi.org/10.1007/s13311-020-00832-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00832-7