
Abstract
Pathogenic variants in KCNT1 represent an important cause of treatment-resistant epilepsy, for which an effective therapy has been elusive. Reports about the effectiveness of quinidine, a candidate precision therapy, have been mixed. We sought to evaluate the treatment responsiveness of patients with KCNT1-related epilepsy. We performed an observational study of 43 patients using a collaborative KCNT1 patient registry. We assessed treatment efficacy based upon clinical seizure reduction, side effects of quinidine therapy, and variant-specific responsiveness to treatment. Quinidine treatment resulted in a > 50% seizure reduction in 20% of patients, with rare patients achieving transient seizure freedom. Multiple other therapies demonstrated some success in reducing seizure frequency, including the ketogenic diet and vigabatrin, the latter particularly in patients with epileptic spasms. Patients with the best quinidine response had variants that clustered distal to the NADP domain within the RCK2 domain of the protein. Half of patients did not receive a quinidine trial. In those who did, nearly half did not achieve therapeutic blood levels. More favorable response to quinidine in patients with KCNT1 variants distal to the NADP domain within the RCK2 domain may suggest a variant-specific response.
Similar content being viewed by others


Introduction
Pathogenic variants in the potassium channel gene KCNT1 have emerged as an important cause of epilepsy with a wide phenotypic spectrum, including autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) in children and adults, an early-onset epileptic encephalopathy (EOEE) in infants and children, and the most severe form, epilepsy of infancy with migrating focal seizures (EIMFS) in neonates and infants [1,2,3,4]. The epilepsy in these patients, particularly those with an EIMFS or EOEE phenotype, is often highly refractory and is accompanied by severe developmental and functional disability. Patients with KCNT1-related epilepsy typically respond poorly to treatment with conventional antiseizure medications, further impairing their quality of life.
Recently, the antiarrhythmic and antimalarial agent quinidine, a sodium and potassium channel blocker, has emerged as a potential precision therapy for KCNT1-related epilepsy [5, 6]. In vitro functional assays have demonstrated mutant gain-of-function effects relative to the wild-type channel that can be reversed by quinidine, and initial case reports in patients showed a significant reduction in seizure burden [7, 8]. However, subsequent reports demonstrated less favorable results, raising questions about quinidine’s role in treating KCNT1-related epilepsy [9,10,11,12,13,14].
We sought to evaluate the treatment responsiveness of patients with KCNT1-related epilepsy to conventional therapies, as well as quinidine, in order to better inform treatment of this patient population with highly refractory epilepsy. We find that response rates to quinidine were low overall. A variety of other treatments were reported as helpful for some patients. A significant subset of patients in our cohort did not receive quinidine, and in those who did, there was variability in maximum blood levels achieved, suggesting that the true effect of this precision medicine therapy may not be accurately reflected in our observational data.
Methods
Institutional review boards at the Children’s Hospital of Philadelphia and the Danish Epilepsy Centre approved this study. Informed consent was obtained from the parents or legal guardian of each patient following local institutional review board requirements. Patients were enrolled in a collaborative KCNT1 registry at the Children’s Hospital of Philadelphia and the Danish Epilepsy Centre by caregivers and/or treating clinicians who self-referred to the registry. The registry prospectively enrolls all patients with KCNT1-related epilepsy, regardless of clinical phenotype. Those patients reported in this paper are new cases that have not been previously published. All KCNT1 variants were identified through clinically based testing, and were reviewed by a genetic counselor and neurogeneticist with expertise in KCNT1-related epilepsy to confirm pathogenicity. Subjects with reported KCNT1 variants that were unlikely to be pathogenic were excluded from analysis. Classification of phenotype (EIMFS, EOEE, ADNFLE) was made by the treating clinicians during usual clinical care and confirmed by a neurologist with expertise in KCNT1-related epilepsy. Where discrepancies arose between the reported phenotype and the clinical information obtained (for example, EIMFS with report of first seizure after 1 year of age), further information was sought to clarify the phenotype and subjects were reclassified if appropriate. Decisions regarding the choice in antiseizure therapies were made by the patient’s neurologist. After enrollment, clinical information was elicited from caregivers and clinicians via online questionnaire and supplemented by medical records when available. Data collected include demographic information, clinical characteristics, age of epilepsy onset, seizure semiology and frequency, current and past antiseizure therapies, and assessment of treatment efficacy with commonly used antiseizure medications, vagal nerve stimulator, the ketogenic diet (KD), and quinidine. Caregivers and clinicians were asked which treatment option led to the best seizure control. In those patients who received quinidine, percent reduction in clinically observed seizures was reported. As some responses to quinidine could be transient, percent reduction in seizures was considered sustained if it persisted for at least 3 months. Success rates for each therapy were calculated, which represent the number of caregivers or clinicians rating a certain treatment as the most efficacious compared with the total number of patients who attempted that particular therapy.
Data are summarized using descriptive statistics, including median and interquartile ranges for continuous variables and percentages for categorical variables. Comparisons between groups were made using Fisher’s exact test for categorical exposure variables, Wilcoxon–Mann–Whitney test for continuous exposure variables with nonparametric distributions, and ANOVA with Tukey’s post hoc test for continuous exposure variables with normal distributions.
Results
The collaborative KCNT1 registry contains data on 43 patients with KCNT1-related epilepsy. To our knowledge, this represents the largest cohort of patients with KCNT1-related epilepsy to date. The majority of patients had a phenotype consistent with epilepsy of infancy with migrating focal seizures (EIMFS, 28/43 patients; 65%). The remaining patients had clinical phenotypes consistent with an early-onset epileptic encephalopathy (EOEE, 9/43 patients; 21%) or autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE, 6/43 patients; 14%). Table 1 describes the clinical characteristics of this cohort, stratified according to clinical phenotype. Overall, patients with KCNT1-related epilepsy experienced multiple seizure types that were intractable to therapy, with the majority of patients (65%) experiencing seizures on a daily or near-daily basis. More specifically, patients with EIMFS demonstrated an earlier age of seizure onset (< 6 months: EIMFS, 97%; EOEE, 55%; ADNFLE, 0%; > 6 months: EIMFS, 4%; EOEE, 44%; ADNFLE, 100%; p = 0.01) and were more likely to report secondarily generalized seizures (EIMFS, 57%; EOEE, 11%; ADNFLE, 17%; p = 0.02), hemiclonic seizures (EIMFS, 64%; EOEE, 22%; ADNFLE, 0%; p = 0.01), and myoclonic seizures (EIMFS, 71%; EOEE, 67%; ADNFLE, 0%; p = 0.01). Epileptic spasms were also frequently reported in patients with EIMFS (36%) and EOEE (56%). There were no statistically significant differences among phenotypes in terms of sex, other reported seizure semiologies, or seizure frequency. There were also no differences in seizure frequency as a function of patient age when considering the cohort as a whole [F(4,38) = 0.92, p = 0.08] or when considering only patients with an EIMFS phenotype [F(4,23) = 0.38, p = 0.69].
A common feature among patients with KCNT1-related epilepsy is their poor response to antiseizure medications. Patients overall were treated with an average of 9 antiseizure therapies (mean, 9.0; SD, 3.6). Those patients with EIMFS were treated with a higher number of therapies than other patients, a difference that achieved statistical significance when comparing EIMFS with ADNFLE patients [mean (SD): EIMFS, 10.2 (3.1); EOEE, 7.7 (3.4); ADNFLE, 5.0 (2.8); F(2,40) = 7.84, p < 0.01; ADNFLE vs EIMFS − 5.21 ± 1.41, p < 0.01]. Table 2 describes the most commonly prescribed antiseizure treatments, stratified according to phenotype. For patients with KCNT1-related EIMFS, the most commonly prescribed therapies were levetiracetam (96%), phenobarbital (96%), ketogenic diet (93%), topiramate (79%), and valproic acid (68%). Treatment approaches for KCNT1-related EOEE were similar and commonly included levetiracetam (100%), topiramate or valproic acid (67% each), and clonazepam, lacosamide, lamotrigine, phenobarbital, or vagal nerve stimulator (44% each). For patients with KCNT1-related ADNFLE, the most common therapies included phenytoin or valproic acid (67% each), followed by lacosamide, lamotrigine, or topiramate (50% each). Quinidine treatment was attempted in 20 patients and, more specifically, in 61% of patients with an EIMFS phenotype and 33% of patients with an EOEE phenotype who had a known variant in KCNT1. Quinidine was not utilized in any patients with an ADNFLE phenotype in our registry.
Given previous reports of variable quinidine efficacy in patients with KCNT1-related epilepsy, we sought to better quantify the reduction in seizure frequency seen with this medication. Table 3 describes the clinical characteristics of the 20 patients in our cohort who received a trial of quinidine. The data listed includes patient age, KCNT1 variant, epilepsy phenotype, previous medications tried, medications used concurrently with quinidine, quinidine response, quinidine dose and maximum blood level (when available), and occurrence of QTc prolongation with quinidine. Because quinidine responsiveness itself could change over time, Fig. 1 depicts the percentage of patients who achieved a particular sustained response to quinidine that lasted at least 3 months. These data demonstrate that approximately half of patients either received no response (8/20, 40%) or reported that seizures worsened (3/20, 15%) despite quinidine therapy. A > 50% reduction in seizures was seen in only 20% of patients (4/20), and sustained seizure freedom was only rarely reported (1/20, 5%, patient 5). The majority of patients reporting seizure freedom initially (2/3, patients 6 and 12) ultimately had recurrence of seizures. There was no statistically significant difference in seizure frequency between patients who received a quinidine trial and those who did not, suggesting that those who received quinidine did not have a more severe phenotype. Side effects attributed to quinidine included prolonged QTc interval (47%), sedation (11%), arrhythmia (5%), elevated liver function tests (5%), and rash (5%). One patient experienced an arrhythmia during quinidine treatment, which consisted of asymptomatic ventricular tachycardia at a quinidine dose of 120 mg/kg/day with a blood level of 1.8. This arrhythmia resolved with initiation of a lidocaine infusion. Given previous reports suggesting that patients younger than 4 years may respond more favorably to quinidine, we evaluated the relationship between age and quinidine responsiveness in our cohort [10]. Our data indicate that the youngest patients are less likely to respond to quinidine compared with older patients (no improvement/worsening: median (IQR), 11 months (4 months, 3 years), vs any improvement: median (IQR), 4 years (3 years, 6 years); Z = − 2.4, p = 0.02).

Sustained efficacy of quinidine in KCNT1-related epilepsy. Response to quinidine was considered sustained if it lasted at least 3 months
We also assessed the maximum blood level achieved in patients treated with quinidine, noting that the anticipated therapeutic range is 2 to 5 μg/mL. For many patients, therapeutic blood levels were not achieved, with 45% of patients failing to reach a blood level of 2.0 (median, 2.0; IQR, 0.6, 2.9). There was no significant difference noted in the maximum blood level achieved when comparing patients who benefited from quinidine with those who did not (no improvement or seizures worsened: median (IQR), 1.9(1.2, 2.5), vs any improvement in seizures: median (IQR), 2.0 (0.6, 5.5); Z = − 0.2, p = 0.85). Because concomitant use of enzyme-inducing medications could lower serum levels of quinidine [15], we evaluated for a relationship between quinidine blood levels and concomitant use of carbamazepine, phenytoin, or phenobarbital. We found no statistically significant difference in maximum blood level achieved when comparing patients concomitantly taking enzyme-inducing antiseizure medications with patients not taking enzyme-inducing medications (nonenzyme inducers: median (IQR), 2.0 (1.8, 2.9), vs enzyme inducers: median (IQR), 0.6 (0.6, 0.6); Z = 1.7, p = 0.1). Notably, this analysis was limited by having only 2 patients with available blood level data in the enzyme-inducing group. Because QTc prolongation could have been a limiting factor in achieving therapeutic blood levels, we assessed the relationship between these parameters. We found no significant difference in the maximum blood level achieved when comparing patients who experienced prolonged QTc with those who did not (normal QTc: median (IQR), 0.65 (0.6, 2.0), vs prolonged QTc: median (IQR), 2.1 (2.0, 2.9); Z = − 1.56, p = 0.12). We also found no significant difference in patient age at the time of quinidine use and the occurrence of QTc prolongation (QTc normal: median (IQR), 1.2 years (0.4 year, 6 years), vs QTc prolonged: median (IQR), 3 years (1.5 years, 3 years); Z = − 0.31, p = 0.76).
Because of the limited efficacy of quinidine in our cohort, we evaluated the efficacy of other more conventional antiseizure treatments. A variety of therapies were rated by caregivers and physicians as the most successful for patients as follows: the ketogenic diet (9/29, 31%), vigabatrin (4/11, 36%), quinidine (3/20, 15%), cannabidiol (2/14, 14%), vagal nerve stimulation (2/14, 14%), phenytoin (2/19, 11%), clobazam (3/31, 10%), clonazepam (2/20, 10%), valproic acid (2/29, 7%), and topiramate (1/31, 3%). Notably, vigabatrin was used in patients with and without epileptic spasms (no spasms, 6/11, 55%; spasms, 5/11, 45%); however, all of the patients (4/4, 100%) for whom it was reported to be the most effective medication had epileptic spasms as a seizure type.
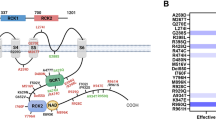
Previous reports of treatment with quinidine in patients with KCNT1-related epilepsy suggested a variable responsiveness to this therapy; therefore, we evaluated whether a variant-specific response to treatment might underlie some of this effect. Figure 2 depicts a schematic of the KCNT1 protein overlaid with the best response achieved by patients with specific variants to quinidine. The important functional domains of the KCNT1 protein, including the RCK1, RCK2, and NADP domains, are noted. From this analysis, several interesting themes emerged. First, 2 of the more commonly occurring variants, G288S and R474H, demonstrate different responses to treatment. Patients with G288S variants showed both responsiveness and nonresponsiveness to quinidine; however, no patients with R474H variants responded to quinidine. Second, the patients who responded best to quinidine, demonstrating at least transient seizure freedom of 1 month, had the following variants: R929Q, R950Q, and R961S. All 3 of these variants are located immediately distal to the NADP domain within the RCK2 domain. This region of the protein is hypothesized to be important in coupling sensitivity to intracellular sodium levels with channel gating [16, 17].

Responsiveness to treatment with quinidine is displayed according to the location of the mutation in the KCNT1 protein. Patients who achieved at least temporary seizure freedom on quinidine lasting > 1 month had mutations that clustered immediately distal to the NADP binding domain within the RCK2 domain. Yellow residues indicate sites with seizure freedom. Purple residues denote sites with responsiveness to quinidine. Red residues denote sites without responsiveness to quinidine. The blue residues denote the locations of the RCK1 and RCK2 domains in the KCNT1 protein, while the green residues denote the location of the NADP binding domain. Patient number (1-20) is listed in parentheses next to responsiveness data. Note: patient 3 had 2 variants in KCNT1 (R428Q and Q651R) and is listed once in the figure
Discussion
This study evaluated the treatment responsiveness of patients with KCNT1-related epilepsy to better inform treatment in this population of patients with refractory epilepsy. There are several clinically meaningful findings. First, quinidine led to a > 50% reduction in seizure frequency in only 20% of patients receiving this therapy, with sustained seizure freedom occurring rarely. Second, a variety of other therapies were reported successful for some patients, with vigabatrin being reported as specifically helpful in patients with epileptic spasms as part of their phenotype. Third, despite treatment with many antiseizure therapies, only half of patients with de novoKCNT1 variants and a resultant epileptic encephalopathy received a trial of quinidine. Fourth, subtherapeutic blood levels of quinidine were often reported. Fifth, the KCNT1 variants that responded best to quinidine therapy clustered distal to the NADP binding domain within the RCK2 domain of the protein.
For the clinician considering the optimal regimen for a patient with KCNT1-related epilepsy, our data indicate that quinidine, the only proposed precision therapy, had limited efficacy, leading to a > 50% reduction in seizures in only 20% of patients. There are a number of possible explanations for this finding. First, quinidine may not penetrate the blood–brain barrier well enough to modulate channel activity in the brain, even at therapeutic serum levels [5]. This notion is supported by a previous study demonstrating that average CSF levels of quinidine in adults were 16% of unbound serum concentrations (range, 4-37%) [18]. We are not aware of any studies of CSF quinidine levels in patients with KCNT1-related epilepsy, particularly those with a more favorable response to the medication. Second, quinidine therapy may be discontinued prematurely, before therapeutic levels can be reached. Our data demonstrate that subtherapeutic blood levels of quinidine were reported in almost half of patients. There are several potential explanations for this finding. One might hypothesize that QTc prolongation would be the most salient limiting factor, as noted in a recent study of adult KCNT1 patients with an ADNFLE phenotype [9]. In our pediatric cohort, we did not observe a difference in blood level or in patient age between patients with QTc prolongation compared with those without. It is unclear whether this indicates that QTc prolongation was not the limiting factor in achieving therapeutic levels, or whether some patients experienced this side effect despite their subtherapeutic levels. Because our registry did not collect information about the clinical response taken to prolongation of the QTc interval, we cannot make any definitive conclusions about this issue. Alternatively, some patients may have failed to achieve therapeutic levels because of interactions with other medications. Hepatic enzyme inducers such as phenobarbital and phenytoin are known to decrease serum levels of quinidine by approximately 50% [15]. We attempted to evaluate this in our cohort, finding no statistically significant difference in blood levels of quinidine as a function of the use of enzyme-inducing medications; however, this analysis is limited by a low number of patients and thus should be interpreted cautiously. Third, a relative lack of familiarity with the dosing and monitoring of quinidine given its infrequent use may have played a role in its limited efficacy. Fourth, only half of patients with a known KCNT1 variant and an epileptic encephalopathy received a therapeutic trial. Possible reasons for not trialing quinidine may include limited access to the medication in some locations, physician unfamiliarity with dosing or drug monitoring, concerns about potential side effects, or concerns about insufficient evidence favoring quinidine treatment. Our data demonstrate that side effects are frequent, particularly QTc prolongation, though only 1 patient in our cohort developed a reversible cardiac arrhythmia requiring discontinuation of quinidine. All of these potential issues may limit our estimates of the effectiveness of quinidine in KCNT1-related epilepsy.
Whereas previous studies have suggested that patients who are younger than 4 years of age at the time of their quinidine trial may respond more favorably to quinidine, we were unable to replicate that result [10]. In our cohort, the patients who responded the best to quinidine, patients 5, 6, and 12, were aged 6 years, 2 years, and 12 months, respectively. Our oldest patient, patient 16, experienced a > 50% in seizures despite starting quinidine at age 14 years. It is possible that age may be 1 factor in determining a patient’s likelihood to respond to quinidine; however, there may be other poorly understood factors influencing treatment response that are challenging to evaluate in relatively small cohorts of patients such as this. It is possible that some component of the variable response to quinidine is related to a true variant-specific response to the medication. Whereas in vitro studies have shown a reduction in channel current in the presence of quinidine, not all variants responded to the same degree, supporting the notion that quinidine may be more effective for some variants than others [7, 11]. While our results should be interpreted as preliminary, it is intriguing that the patients who responded the best to quinidine in our cohort had variants located immediately distal to the NADP binding domain within the RCK2 domain of the protein. Previous work has demonstrated that binding of NAD+ or NADP+ increases the affinity of the channel for Na+, leading to increased channel opening [16]. Other work has demonstrated that a sodium-sensitive motif exists within the RCK2 domain that is important for coupling sodium sensitivity to channel gating [17]. How quinidine may be interfacing with the channel to reduce conductance is currently unknown. It will be important for future studies to evaluate the impact of these variants on channel conductance and quinidine response in vitro. It will also be important, ultimately, to correlate in vitro quinidine responsiveness with CSF quinidine levels in patients who respond to treatment. One might expect that a positive response to treatment could be achieved in patients with more quinidine-sensitive variants at lower serum/CSF concentrations compared with patients possessing less sensitive KCNT1 variants.
Our study has some noteworthy limitations. First, our measures of treatment responsiveness are based upon the reduction in clinically apparent seizures reported by caregivers and clinicians. Clearly, this method of self-report is subject to bias, especially pertaining to recall and placebo effect. As none of the treatment options investigated in our cohort were superior in their reported effectiveness, these biases are unlikely to be falsely identifying 1 therapy as superior. Whereas electroencephalographic seizure burden assessment may be a desirable measure of treatment responsiveness, it is challenging in an international registry of patients with rare diseases. Given that everyday management of patients with intractable epileptic encephalopathies is heavily based upon reports of clinical response, we believe that our data provide useful information for clinicians despite this limitation. Second, our measurement of the success rate of treatments other than quinidine is suboptimal. We provide these data as success rates because our registry does not quantify percent seizure reduction with all possible therapies. Despite the limitations of this measure, we consider these data important to report, especially in light of the limited response to quinidine in our cohort. Third, our registry is not sufficiently powered to determine whether there is a true difference in the responsiveness of individual KCNT1 variants to treatment with multiple potentially effective agents. Fourth, we can only speculate based on our data regarding the possible reasons for the limited responsiveness of KCNT1 patients to quinidine. Several of our analyses are limited by low numbers of patients, which may be an additional factor limiting assessments of quinidine response. Ultimately, a blinded and controlled trial of quinidine in the pediatric population is needed to evaluate the efficacy of quinidine in KCNT1-related epilepsy. Such a trial would mitigate many of the sources of bias inherent in our study. To date, the only controlled trial of quinidine was performed in adult patients by Mullen and colleagues. This trial demonstrated that quinidine was not effective for reducing seizure burden and that QTc prolongation was a significant, dose-limiting side effect. However, the trial was limited by small sample size and by failure to achieve therapeutic serum levels in several patients because of side effects [9]. This high risk of toxicity underscores the importance of finding a safer, more effective therapy for patients with KCNT1-related epilepsy,
In summary, our data demonstrate that multiple treatments may be helpful in reducing seizure frequency for patients with KCNT1-related epilepsy, though we cannot establish a clearly superior treatment for this patient population as a whole. Quinidine efficacy was limited, though our data do not definitely establish the reasons for this low response rate. Until a precision therapy is developed with an improved side effect profile, thoughtful consideration of the potential benefits and risks of a trial of quinidine is reasonable in selected patients with KCNT1-related epileptic encephalopathy. When initiating quinidine treatment in this population, we suggest a protocol-based approach to maximize serum levels while monitoring for side effects. Additional treatment considerations in this patient population are several, including the ketogenic diet or vigabatrin, the latter particularly in patients with epileptic spasms as part of their phenotype.
References
Moller RS, Heron SE, Larsen LHG, Lim CX, Ricos MG, Bayly MA, et al. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia. 2015;56:e114–20.
Ishii A, Shioda M, Okumura A, Kidokoro H, Sakauchi M, Shimada S, et al. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene. 2013;531:467–71.
Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. Nature Publishing Group; 2012;44:1188–90.
Barcia G, Fleming MR, Deligniere A, Gazula V-R, Brown MR, Langouet M, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44:1255–9.
Grace AA, Camm AJ. Quinidine. Wood AJJ, editor. N Engl J Med. Massachusetts Medical Society; 1998;338:35–45.
Tamargo J, Caballero R, Gómez R, Valenzuela C, Delpón E. Pharmacology of cardiac potassium channels. Cardiovasc Res. Oxford University Press; 2004;62:9–33.
Milligan CJ, Li M, Gazina E V, Heron SE, Nair U, Trager C, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol. 2014;75:581–90.
Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol. 2014;76:457–61.
Mullen SA, Carney PW, Roten A, Ching M, Lightfoot PA, Churilov L, et al. Precision therapy for epilepsy due to KCNT1 mutations. Neurology. 2018;90:e67–72.
Abdelnour E, Gallentine W, McDonald M, Sachdev M, Jiang Y-H, Mikati MA. Does age affect response to quinidine in patients with KCNT1 mutations? Report of three new cases and review of the literature. Seizure. 2018;55:1–3.
Mikati MA, Jiang Y, Carboni M, Shashi V, Petrovski S, Spillmann R, et al. Quinidine in the treatment of KCNT1-positive epilepsies. Ann Neurol. 2015;78:995–9.
McTague A, Nair U, Malhotra S, Meyer E, Trump N, Gazina E V., et al. Clinical and molecular characterization of KCNT1 -related severe early-onset epilepsy. Neurology. 2018;90:e55–66.
Numis AL, Nair U, Datta AN, Sands TT, Oldham MS, Patel A, et al. Lack of response to quinidine in KCNT1 -related neonatal epilepsy. Epilepsia. 2018.
Chong PF, Nakamura R, Saitsu H, Matsumoto N, Kira R. Ineffective quinidine therapy in early onset epileptic encephalopathy with KCNT1 mutation. Ann Neurol. 2016;79:502–3.
Data JL, Wilkinson GR, Nies AS. Interaction of quinidine with anticonvulsant drugs. N Engl J Med. 1976;294:699–702.
Tamsett TJ, Picchione KE, Bhattacharjee A. NAD+ activates KNa channels in dorsal root ganglion neurons. J Neurosci. 2009;29:5127–34.
Zhang Z, Rosenhouse-Dantsker A, Tang Q-Y, Noskov S, Logothetis DE. The RCK2 domain uses a coordination site present in Kir channels to confer sodium sensitivity to Slo2.2 channels. J Neurosci. 2010;30:7554–62.
Ochs HR, Greenblatt DJ, Lloyd BL, Woo E, Sonntag M, Smith TW. Entry of quinidine into cerebrospinal fluid. Am Heart J. 1980;100:341–6.
Acknowledgments
Steffen Syrbe received funding from a grant from the Dietmar-Hopp-Stiftung (23011236 to S.S.)
• Tracy Gertler received funding from an National Center for Advancing Translational Sciences grant (KL2TR001424 to T.G.)
• Ingo Helbig was supported by intramural funds by the University of Kiel and through grants by the German Research Foundation (DFG, HE5415/5-1, HE 5415/6-1, HE 5415/7-1) within the framework of the EuroEPINOMICS-RES project through the Eurocores program of the European Science Foundation (ESF).
• David Bearden received funding from and served as a consultant for Q-State Biosciences
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they do not have conflicts of interest.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Fitzgerald, M.P., Fiannacca, M., Smith, D.M. et al. Treatment Responsiveness in KCNT1-Related Epilepsy. Neurotherapeutics 16, 848–857 (2019). https://doi.org/10.1007/s13311-019-00739-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00739-y