
Abstract
Cholesterol 24-hydroxylation is the major mechanism for cholesterol removal from the brain and the reaction catalyzed by cytochrome P450 46A1 (CYP46A1), a CNS-specific enzyme. This review describes CYP46A1 in the context of cholesterol homeostasis in the brain and summarizes available experimental data on CYP46A1 association with different neurologic diseases, including the mechanisms by which changes in the CYP46A1 activity in the brain could be beneficial for these diseases. The modulation of CYP46A1 activity by genetic and pharmacologic means is also presented along with a brief synopsis of the two clinical trials that evaluate CYP46A1 as a therapeutic target for Alzheimer’s disease as well as Dravet and Lennox–Gastaut syndromes.
Similar content being viewed by others
Introduction
Currently, there are no disease-modifying treatments for Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD) with all of the therapies being symptomatic. Hence, different targets and pathways are now evaluated for development of disease-modifying treatments for these progressive and debilitating conditions. Among them are the studies of cytochrome P450 46A1 (CYP46A1), the cholesterol 24-hydroxylase and important brain enzyme.
Essentials of Cholesterol Homeostasis in the Brain
The brain is rich in cholesterol, a key component of eukaryotic membranes and a precursor of different neurosteroids as well as oxysterols. Cholesterol in the brain is mainly present in the two pools: in myelin sheaths of white matter, which insulate axons and enhance propagation of action potential, and in the membranes of glial cells and neurons of gray matter, which are essential for cognitive functions. In mice, these pools contain ~70% and ~30% of cholesterol, respectively, with the predicted cholesterol distribution in the second pool being ~20% in glial cells and ~10% in neurons (reviewed in Dietschy and Turley [1]). Cholesterol in myelin sheaths is likely metabolically stable, whereas cholesterol in cellular membranes is metabolically active with the overall rate of cholesterol turnover in the CNS being 0.03% per day (~9.1 years) in humans and 0.4% per day (~0.7 year) in mice (reviewed in Dietschy and Turley [2]). Studies in mice indicate that the rates of cholesterol turnover vary with age and cell type and in adult brain could be as high as ~30% per day in a subset of metabolically active neurons and much lower (~0.16% per day) in the remaining cells [3].
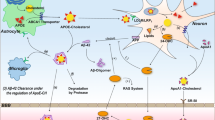
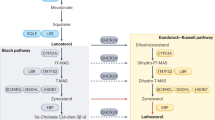
Normally, cholesterol cannot cross the blood–brain barrier; therefore, cholesterol maintenance in the brain includes in situ biosynthesis (almost the only source of cholesterol), cholesterol transport between different cells, and elimination (Fig. 1). Cholesterol biosynthesis, a complex process involving multiple steps, proceeds in the brain via the two major pathways: the Bloch pathway, which is suggested to take place in astrocytes, and the Kandutsch–Russell pathway, which seems to be operative in neurons (reviewed in Pfrieger and Ungerer [4]). Both pathways are important for brain physiology because genetic deficiencies of ∆24-dehydrocholesterol reductase (DHCR24) and 7-dehydrocholesterol reductase (DHCR7) catalyzing the last steps in the Bloch and Kandutsch–Russell pathways, respectively, lead to desmosterolosis and Smith–Lemli–Opitz syndrome, respectively, the rare hereditary diseases characterized by neurological problems [5, 6]. In the brain, the capacity of different cells to synthesize cholesterol varies and depends on age; the glial cells (especially astrocytes) are believed to synthesize most of the brain cholesterol in adults. This cholesterol is then delivered to neurons to satisfy their high demand for cholesterol, probably for building the extensive membrane surface of axons, dendrites, and synapses as well as formation of synaptic vesicles (reviewed in Pfrieger and Ungerer [4] and [7]).

Major pathways of cholesterol homeostasis in adult brain. Arrow thickness indicates the relative quantitative significance of the pathway in humans. Cholesterol (shown as a ball model in orange, the hydroxyl group is in red) is synthesized from acetyl-CoA in both neurons and astrocytes, and then could be effluxed from astrocytes by ABCA1 and transported on high-density lipoprotein (HDL)-like particles to neurons where it is taken up by a receptor-dependent mechanism. Once in neurons, CYP46A1 can convert cholesterol excess to 24-hydroxycholesterol (24HC, shown as a ball model in green, the hydroxyl groups are in red), which could rapidly diffuse to the CSF and then through the blood–brain barrier to the systemic circulation for delivery to the liver. Some of the HDL-like particles could travel to the cerebrospinal fluid (CSF) and then gain access to the systemic circulation via the receptor-mediated endocytosis. A small portion of cholesterol could be esterified by sterol-O-acetyltransferase 1 (SOAT1) to form cholesterol esters (CE)
To deliver cholesterol to neurons, astrocytes synthesize apolipoproteins, mainly E (APOE), J (APOJ or clusterin), and D (APOD), which after secretion serve as acceptors for astrocytic cholesterol effluxed by the ABC transporters (mainly by ABCA1). The secreted APOE and APOJ could bind cholesterol and other lipids to form the HDL-like lipoprotein particles, whereas APOD binds lipids and associates with other lipoproteins (reviewed in Pfrieger and Ungerer [4], [8], [9], and Elliott et al. [10]). The astrocyte-produced apolipoprotein particles reach neurons and other cells and bind to the cell surface receptors. The prototypic low-density lipoprotein receptor (LDLR, expressed on both glial and neuronal cells) and the LDLR-related protein 1 (LRP1, predominantly expressed in neurons) are the two major receptors that are probably involved in cholesterol uptake by brain cells. The brain also expresses other receptors, including the very-low-density lipoprotein receptor (VLDLR), the APOE receptor 2 (APOER2 or LRP8), and megalin (LRP2), that can bind lipoprotein particles (reviewed in Pfrieger and Ungerer [4], Arenas et al. [11], and Bjorkhem and Meaney [12]). Remarkably, recently, an individual with a total lack of APOE was identified and found to have normal cognitive and neurological functions as well as normal brain structure on magnetic resonance imaging [13]. This clinical finding suggested that the APOE functions are redundant and other apolipoproteins in the brain can compensate for APOE absence.
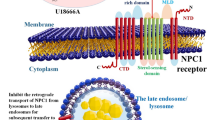
Once taken up by a cell, cholesterol is delivered to late endosomes/lysosomes, in which the Niemann-Pick C1 (NPC1) and C2 (NPC2) proteins bind cholesterol and mediate its export to endoplasmic reticulum and plasma membranes. The importance of the endolysosomal cholesterol traffic in the brain is highlighted by the mutations in NPC1 and NPC2, which lead to Niemann-Pick type C disease. This is a lysosomal storage disorder characterized by cholesterol accumulation in the endolysosomes of all the tissues with, however, the most devastating consequence of this accumulation being in the brain, which develops neurodegeneration and neuronal abnormalities (reviewed in Vance and Karten [14]).
To maintain the steady-state levels, cholesterol biosynthesis in the brain is balanced by cholesterol elimination. One mechanism is cholesterol hydroxylation by the cytochrome P450 enzymes: ubiquitous CYP27A1 [15], steroidogenic CYP11A1 [16], and CNS-specific CYP46A1 [17], which convert cholesterol to 27-hydroxycholesterol, pregnenolone, and 24S-hydroxycholesterol (24HC), respectively. Of these enzymatic reactions, cholesterol 24-hydroxylation by CYP46A1 is quantitatively significant and was found to account for elimination of 75–85% and 40–50% of cholesterol excess from human and mouse brains, respectively [18, 19]. Unlike membrane-impermeable cholesterol, 24HC could rapidly diffuse out of cells and gain access to the systemic circulation (~98%) or cerebrospinal fluid (CSF, ~2%) for delivery to the liver for further degradation to bile acids [20, 21]. CYP46A1 is a microsomal enzyme, expressed preferentially in the brain and to a much lower extent in the retina [17, 22, 23]. In the brain, CYP46A1 is found in pyramidal neurons of the hippocampus and cortex, and Purkinje cells of the cerebellum as well as hippocampal and cerebellar interneurons [24,25,26]. In both humans and mice, the brain CYP46A1 levels rapidly increase after birth and reach a steady state by 1 year in humans (and 2–4 weeks in mice) [17], suggesting CYP46A1 importance for brain functions in adults. Studies in mice indicate that changes in the rate of cholesterol 24-hydroxylation lead to compensatory decreases or increases in the rate of cholesterol biosynthesis by yet an unknown mechanism, and thereby suppress or stimulate brain cholesterol turnover [18, 27,28,29]. Because of the high CYP46A1 expression in the brain, serum 24HC levels were found to be a biomarker of the enzyme activity in the brain and reflect the ratio of the brain weight to liver volume [17, 30]. Apolipoprotein-mediated transport through the CSF is the other mechanism by which the brain clears cholesterol excess. If cholesterol 24-hydroxylation removes most and approximately a half of cholesterol excess from human and mouse brains, respectively [18, 19], then apolipoprotein-mediated transport through the CSF could lead to up to 15–25% and 50–60% of the brain cholesterol elimination in humans and mice, respectively. The details of this mechanism are still under investigation but likely involve apolipoprotein A1 (APOA1) as APOA1-containing particles could diffuse to the CSF and then be taken up via endocytosis by the receptors (LRP1 and scavenger receptor class B type 1) that are expressed on the brain capillary endothelial cells (reviewed in Pfrieger and Ungerer [4] and Zhang and Liu [31]). In addition to cholesterol elimination, a small excess of brain cholesterol is esterified by ACAT1 (acyl-coenzyme A:cholesterol acyltransferase 1) or SOAT1 (sterol O-acyltransferase 1), which resides in the endoplasmic reticulum and acts on intracellular cholesterol ( [32, and reviewed in Chang et al. [33]). LCAT (lecithin:cholesterol acyltransferase) is another cholesterol-esterifying enzyme in the brain, which is secreted by astrocytes and esterifies cholesterol on nascent APOE-containing particles [34]. In healthy adult humans and mice, cholesterol esterification is of very minor significance because cholesterol esters constitute only about 1% of total brain cholesterol (reviewed in Bjorkhem and Meaney [12]). Thus, of the mechanisms which target cholesterol excess in the brain, cholesterol removal by CYP46A1 is of key importance; this mechanism represents not only the major pathway of cholesterol elimination but also the means to control brain cholesterol biosynthesis and turnover.
CYP46A1 Activity Modulation for Major Diseases of the Brain
This section will describe the three major mechanisms by which modulation of CYP46A1 activity in the brain could affect brain processes and function, namely by changing: 1) the steady-state levels of cholesterol; 2) the levels of 24HC, a biologically active molecule; and 3) the rate of brain cholesterol turnover (Fig. 2).

Major mechanisms whereby modulation of CYP46A1 activity can affect brain processes. The three major mechanisms are changes in the brain cholesterol levels, 24-hydroxycholesterol (24HC) levels, and cholesterol turnover. The brain cholesterol levels could modulate the progression of several neurodegenerative disorders. The 24HC cholesterol levels can affect the activation of N-methyl-d-aspartate receptors (NMDARs) involved in memory and cognition and the activation of liver X receptors (LXRs), important transcription factors controlling gene expression in multiple pathways. The brain cholesterol turnover can alter physicochemical properties of plasma and subcellular membranes and thereby synaptogenesis and synaptic processes as well as lead to lipid raft rearrangement. The latter can in turn alter phosphorylation and amyloid β production. In addition, the brain cholesterol turnover can determine the rate of cholesterol biosynthesis and hence the production of non-sterol isoprenoids required for protein prenylation. The question marks indicate the lack of sufficient experimental evidence
CYP46A1 and Brain Cholesterol Levels in AD, HD, and PD
CYP46A1 was first linked to the diseases of the brain by the measurements of plasma 24HC levels. The levels of this sterol were slightly increased in the plasma of patients with early AD and non–AD demented patients but were unchanged in patients with depression [35]. Conversely, patients with advanced AD had decreased 24HC levels in their plasma, likely reflecting the marked destruction of the CNS and hence a reduction in the CYP46A1 levels and the flux of 24HC from the brain [36, 37]. In agreement with this explanation, the measurements of 24HC in the four brain regions (the frontal and occipital cortexes, basal ganglia, and pons, Fig. 3) of patients with AD revealed decreased sterol levels in all four regions and a close coupling between cholesterol biosynthesis and elimination [38]. Simultaneously, the brain of patients with AD was discovered to have a different CYP46A1 distribution as compared to that in control subjects; in addition to neurons, some of the astrocytes in the AD-affected brain were found to express CYP46A1 [24, 39]. Because AD leads to synaptic and then neuronal loss and thereby a decrease in the CYP46A1 content, CYP46A1 expression in astrocytes was suggested to compensate for the CYP46A1 decrease due to neurodegeneration [39]. Notably, CYP46A1 was revealed to be a highly polymorphic gene with the frequency of some of its intronic polymorphism being as high as 40% (https://www.ncbi.nlm.nih.gov/snp/). Some but not all genetic studies linked the CYP46A1 intronic polymorphisms to AD (reviewed in Russell et al. [26]), yet the current meta-analyses suggest that only some of the CYP46A1 polymorphisms could be associated with AD albeit being minor risk factors and ethnicity specific [40,41,42].

Brain regions affected by Alzheimer’s, Huntington’s, and Parkinson’s diseases. The regions in color on sagittal sections are those that are mostly affected by the disease; the regions in gray are those that are affected to a lesser extent or at a later disease stage. Upward arrows, downward arrows, and left-right arrows indicate increase, decrease, and no change, respectively, in the levels of cholesterol (C, in orange) and 24-hydroxycholesterol (24HC, in green), and CYP46A1 (in magenta) in different regions of the disease-affected brain as discussed in the main text
Unfortunately, there have been no systematic investigations of the absolute cholesterol levels in different brain regions of patients with AD. Available data are scarce and equivocal [43, 44]. Perhaps the most comprehensive was the study on AD samples that evaluated the frontal and occipital cortexes, basal ganglia, and pons not only for the levels of different sterols but also for cholesterol content, which was increased only in the basal ganglia [38]. In a different study, cholesterol was found to be increased in the gray matter frontal cortex of the AD carriers of the APOE4 genotype as compared to the APOE4 control subjects [45]. Conversely, the cholesterol levels in crude hippocampal membranes from the nongenotyped AD patients were reported to have only minor changes [46]. Consistent with this report are the data on unchanged cholesterol levels in the temporal and frontal cortexes affected by AD, and decreased cholesterol levels in the lipid raft fraction from the temporal but not frontal cortex [47]. Furthermore, local cholesterol accumulation was detected in the amyloid β-positive presynaptic nerve terminals from the AD cortex [48] as well as in the core of mature, but not diffuse, amyloid plaques [49]. In contrast, cholesterol levels in white matter were found to be reduced in AD patients, likely as a result of extensive axonal demyelination [50]. Thus, available literature data suggest that there are probably region- and membrane-specific changes in the brain cholesterol levels in AD. Also, epidemiologic studies established that the ε4 allele of APOE and variants of APOJ are risk factors for late-onset AD [51,52,53,54], which in part could be due to the altered capacity of the brain apolipoprotein particles to transport and deliver cholesterol (reviewed in Mahley and Rall [55], Mahley [56], and Bu [57]). Accordingly, increase in the CYP46A1 expression or activity in the brain may be beneficial for AD to compensate for changes in the brain apolipoprotein-mediated transport.
HD is an autosomal dominant neurodegenerative disorder due to the mutation in the huntingtin gene [58]. This disease is characterized by cell loss and astrogliosis in several brain regions with the most prominent alterations occurring in the striatum (comprised of caudate nucleus and putamen, Fig. 3), ultimately leading to striatal atrophy [59]. Plasma 24HC levels were shown to be reduced in HD patients and correlate with disease severity and markers of disease progression [60,61,62]. This reduction is consistent with the reported decreases in the 24HC levels in the caudate and putamen as well as the reduction of the CYP46A1 levels in the putamen of the HD patients [63, 64]. Also, of the four studied regions in HD samples (putamen, caudate, gray cortex, white cortex, and cerebellum), only the putamen was found to have increased cholesterol levels as well as the levels of the major cholesterol precursors [64]. This is in contrast to cholesterol increase in the caudate of HD patients in a different study [65], and the suggested impairment in cholesterol biosynthesis in the cortex and striatum of HD patients in a study that quantified the levels of HMGCR, CYP51, and DHCR7 involved in cholesterol biosynthesis [66]. Thus, available data are not consistent; nevertheless, they all suggest that cholesterol homeostasis is altered in the HD-affected brain and that CYP46A1 could be a reasonable therapeutic target for this disease.
Finally, PD, a common neurodegenerative disorder, is characterized by a marked loss of dopaminergic neurons in the pars compacta of the substantia nigra (a region of the midbrain, Fig. 3) as well as the formation of Lewy bodies, which are composed of protein aggregates, mainly those of α-synuclein (reviewed in Lee and Trojanowski [67]). So far, three studies measured the 24HC levels in PD patients. In one study, no changes in plasma levels of total 24HC were found in patients with PD, thus suggesting that CYP46A1 is not affected in this disorder. Interestingly, there was a significant correlation between the levels of 24HC in the patient CSF and disease duration, raising a possibility that this CSF oxysterol may be of value to follow the disease progression [68]. In a different study, which only measured the plasma levels of unesterified 24HC, the PD group had a decrease in the plasma content of this oxysterol [69]. The third study showed that plasma levels of esterified 24HC are significantly reduced in patients with PD and could reflect an impaired catalytic activity of LCAT as result of a higher level of oxidative stress in the CNS. Esterified 24HC in plasma was suggested to be a novel biomarker of neurodegeneration in PD as well as a measure of disease severity and progression [70]. In addition, several brain regions in patients with PD were characterized by a comprehensive lipidomics analysis: these were the primary visual cortex (a region that is mainly devoid of structural but not functional changes) and the amygdala and anterior cingulate cortex (two regions which contain Lewy bodies at the different disease stages but do not have such severe degeneration as the substantia nigra) [71]. In the PD samples, cholesterol levels were only increased in the visual cortex but not amygdala and anterior cingulate cortex. Similarly, the visual cortex had an increase in the 24HC levels and an increase in the levels of CYP46A1 [71]. Lastly, both statins (inhibitors of cholesterol biosynthesis) and higher serum cholesterol were suggested to reduce PD risk, although for both conflicting findings were reported [72,73,74,75,76,77,78]. A recent meta-analysis showed that a protective effect of statins was observed in studies that did not adjust for serum cholesterol and was not observed in studies that performed this adjustment [73]. Additional investigations are required to make definite conclusions about the benefit of CYP46A1 activity modulation in PD.
Brain Effects of 24HC
Modulation of CYP46A1 activity changes the brain levels of 24HC (Fig. 2), a known activator/modulator of at least two receptors—liver X receptors (LXRs) and N-methyl-d-aspartate receptors (NMDARs). LXRs are the transcription factors and master regulators of cholesterol elimination; they are also involved in the regulation of fatty acid and triglyceride biosynthesis and glucose metabolism as well as immune-inflammatory responses (reviewed in Kalaany and Mangelsdorf [79], Glass and Ogawa [80], and Calkin and Tontonoz [81]). LXR activation by synthetic LXR agonists was shown to be neuroprotective and anti-inflammatory in mouse models of AD, PD, traumatic brain injury, stroke, and amyotrophic lateral sclerosis (reviewed in Courtney and Landreth [82]). However, there is no direct evidence that LXRs are activated by 24HC in the brain as the brain expression of LXR target genes was not increased in heterozygous transgenic mice, which overexpressed CYP46A1 and had about a 2-fold increase in their brain 24HC levels [83]. Possibly, a higher than a 2-fold increase in the 24HC levels is necessary to activate LXRs in the brain. Alternatively, rapid diffusion of the 24HC surplus out of cells [21] precludes the oxysterol binding to LXRs, which mainly reside in the nucleus but in some cell types can also be found in the cytoplasm or plasma membranes [84, 85].
24HC is a positive allosteric modulator of NMDARs, which mediate excitatory neurotransmission throughout the CNS and are crucial for synaptic plasticity and learning [86,87,88]. Specifically, 24HC was found to act mainly on NMDARs which contain the GluN2B subunit [89], an important target for NMDAR modulation by ketamine and memantine, the latter being a medication approved by the FDA to ameliorate the symptoms of AD [90, 91]. An increase in 24HC as a result of CYP46A1 activation could enhance the Ca2+ current through NMDARs and improve memory and learning. Also, CYP46A1 activation could be useful when there is a decrease in glutamatergic synaptic transmission, for example in schizophrenia or in overdose with ketamine and phencyclidine, NMDAR antagonists and recreational drugs (reviewed in Xia et al. [92] and Coyle [93]). However, overstimulation of NMDARs leads to excitotoxicity and neuronal cell death and was found in such pathologic conditions as epilepsy and hyperalgesia (reviewed in Collingridge et al. [86]). Accordingly, CYP46A1 should be inhibited in the conditions of NMDAR overstimulation. Of importance is that excessive stimulation of glutamate receptors in mice was shown to induce a loss of membrane cholesterol due to metabolism by CYP46A1 [94], thus creating a vicious cycle of NMDAR overstimulation. Remarkably, in vitro, CYP46A1 was also shown to be activated by glutamate, raising a possibility of a reciprocal relationship between CYP46A1 activity and neurotransmission: increased CYP46A1 activity enhances neurotransmission, and excitatory neurotransmission in turn increases CYP46A1 activity [95]. In vivo studies are required to test this possibility and clearly establish the role of 24HC. Unfortunately, there have yet been no in vivo investigations unambiguously showing uncoupling between changes in the brain 24HC levels and cholesterol turnover, i.e., when the 24HC levels were changed but the rate of cholesterol turnover remained unaffected as indicated by unchanged levels of lathosterol and desmosterol (as markers of the neuronal and astrocytic cholesterol biosynthesis, respectively, reviewed in Pfrieger and Ungerer [4]) and either unchanged or reduced levels of the brain total cholesterol.
Brain Effects of the Altered Cholesterol Turnover
Similar to studies of the individual 24HC effects, the investigation of the individual effects of brain cholesterol turnover is difficult because of the tight link to the 24HC levels. Hence, this section will only consider how cholesterol turnover could affect the membranes and membrane-associated processes in the brain (Fig. 2) by changing the flux of cholesterol and 24HC through the membranes and lipid rafts in the regions where CYP46A1 is expressed. Cholesterol is abundant in plasma membranes, where it regulates the physicochemical properties of the membranes (e.g., membrane fluidity, thicknesses, and permeability). Also, cholesterol serves as a glue for the organization of the membrane microdomains called lipid rafts, which are important for cellular signaling and function of a number of membrane proteins (reviewed in Sezgin et al. [96] and Grouleff et al. [97]).
Perhaps the most prominent effect of the cholesterol turnover could be on synaptic processes and higher-order brain functions; cholesterol was shown to be important for synaptogenesis, virtually all aspects of synaptic transmission (neurotransmitter release, synaptic vesicle traffic, and postsynaptic receptor clusterization) as well as synaptic plasticity [98,99,100,101,102]. This effect will be discussed in the next sections, which will summarize the key findings from the characterizations of mice with altered cholesterol turnover. In addition, changes in the brain cholesterol turnover could lead to lipid raft rearrangement and subsequent alteration of protein phosphorylation and generation of amyloid β. The former point is indirectly supported by a study of the brain phosphoproteome of Cyp46a1−/− mice, which showed a general decrease in protein phosphorylation [103], possibly as a result of altered targeting of protein kinases to their protein substrates and vice versa protein substrates to their kinases, which reside in lipid rafts. The latter point is supported by enrichment of lipid rafts with amyloid precursor protein and the secretases that cleave this protein and generate amyloid β peptides. There are numerous cell culture studies, which link membrane cholesterol and the amyloid β generation, yet in vivo effects of the brain cholesterol turnover have not yet been demonstrated, except only in one study, which convincingly related cholesterol turnover to protein prenylation [104] as described below.
Modulation of CYP46A1 Activity by Genetic Means
CYP46A1 transcription was found to be insensitive to major regulatory axes, except oxidative stress [105]. Studies show that CYP46A1 may be controlled epigenetically [106,107,108] and by the specificity (Sp) transcription factors as the levels of basal expression [109, 110]. So far, the modulation of CYP46A1 activity at the gene level was by Cyp46a1 ablation or CYP46A1-containing adenovirus injections (Table 1).
The generation of Cyp46a1−/− mice was the first genetic manipulation of CYP46A1 activity in mammals [18]. These mice provided seminal knowledge of a role of CYP46A1 in cholesterol homeostasis in the brain and linked cholesterol 24-hydoxylation and cholesterol turnover in the brain to the higher-order brain functions [18, 26, 104, 118, 119]. A lack of CYP46A1 was discovered to lead to severe deficiencies in spatial, associative, and motor learning as well as in hippocampal long-term potentiation [104]. The underlying reason for cognitive deficits was established as well, namely a reduced production of geranylgeraniol, a nonsterol isoprenoid required for hippocampal long-term potentiation [104, 118], synaptic basis for learning and memory. The steady-state cholesterol levels were unchanged in the brain of Cyp46a1−/− mice because the rate of cholesterol biosynthesis was reduced almost 2-fold to compensate for Cyp46a1 ablation [18]; hence, the production of nonsterol isoprenoids was also reduced because these compounds are generated during cholesterol biosynthesis.
The cortical and hippocampal injections of the CYP46A1-containing adenovirus to APP23 or APP/PS mice, the models of AD, were the first genetic manipulation which enhanced CYP46A1 activity in mammals [112]. These injections, before and after the onset of amyloid plaques, increased CYP46A1 expression and the levels of 24HC in the brain, while reducing the amyloid β pathology and rescuing some cognitive deficits [112]. This work provided the first experimental evidence that CYP46A1 could be a therapeutic target for AD. Simultaneously, genetic ablation of Acat1 (or Soat1) in triple transgenic mice, which contained mutant human amyloid precursor protein, presenilin 1, and Tau protein, was found to also increase the 24HC levels, while reducing the cholesterol biosynthesis rate and amyloid pathology [32]. Then, hetero- and homozygous transgenic mice, which overexpressed CYP46A1, were generated [27, 83]. The 24HC levels and the levels of lanosterol, a cholesterol precursor, were increased in these animals indicating enhancement of cholesterol turnover. Notably, homozygous CYP46A1 transgenic mice (15-month-old females) showed an increased expression of the studied pre- and postsynaptic proteins and improvement in spatial memory as assessed by the Morris water maze (MWM) test. This study demonstrated that increased CYP46A1 activity and brain cholesterol turnover are of benefit for cognitive function under normal conditions [27].
In the more recent investigations, additional insights into the role of CYP46A1 activity were obtained. When THY-Tau22 mice, an AD model of tau but not amyloid pathology, received bilateral hippocampal injections of the CYP46A1-containing adenovirus, the CYP46A1 expression was selectively enhanced in the hippocampal neurons. These injections rescued mouse cognitive deficits as well as impaired long-term depression and spine defects but did not affect Tau phosphorylation and the associated gliosis. The authors suggested that CYP46A1 may be a relevant therapeutic target for taupathies, especially in AD [111]. Similarly, there was a beneficial effect of the adenoviral injections of CYP46A1 in the striatum of the R6/2 mice, a HD model. These injections decreased neuronal atrophy; the number, intensity level, and size of huntingtin aggregates; and ameliorated motor deficits. Conversely, when wild-type mice received striatal injections of the virus encoding the short hairpin (sh) Cyp46a1 RNA to reduce the gene expression, they developed the HD phenotype with spontaneous striatal neuron degeneration and motor deficits. Collectively, these and other data provided experimental evidence that CYP46A1 could be a therapeutic target for HD [63]. Yet, when the shCyp46a1 RNA was delivered to the hippocampus of wild-type mice, the cholesterol content was increased in neurons followed by cognitive deficits, enhanced production of the amyloid β peptides, abnormal tau phosphorylation, and hippocampal atrophy due to apoptotic neuronal death [114]. In a different study, which also utilized hippocampal injections of the shCyp46a1 RNA to wild-type mice, similar results were obtained, and, in addition, it was shown that silencing Cyp46a1 expression induces neuronal sclerosis and epileptic activity in mouse hippocampus [115]. Thus, modulation of CYP46A1 activity by genetic means demonstrated that region-specific decreases of CYP46A1 activity may lead to different pathologic consequences (AD- and HD-like pathology and epilepsy) and pointed to the diseases, which may utilize CYP46A1 as a therapeutic target.
Pharmacologic Modulation of CYP46A1 Activity
The CYP46A1 potential as a pharmacologic target was first implicated by the biochemical characterizations of the purified recombinant enzyme [120]. In addition to hydroxylating cholesterol and 24HC, CYP46A1 was found to metabolize structurally diverse C21- and C27-steroids as well as some marketed drugs [120]. These data suggested that CYP46A1 has a flexible active site and prompted structural studies of the enzyme along with the screening of the marketed drugs for the effect on CYP46A1 activity in vitro. Initially, substrate-free and cholesterol substrate–bound crystal structures of CYP46A1 were determined, which confirmed conformational flexibility of the enzyme active site (Fig. 4) [121]. Then, based on the results of drug screening, CYP46A1 was additionally cocrystallized in complex with seven other compounds (Fig. 4), mainly CYP46A1 inhibitors and the FDA-approved pharmaceuticals: the anticonvulsant thioperamide; the antidepressants tranylcypromine and fluvoxamine; the antifungals clotrimazole, posaconazole, and voriconazole; as well as the antineoplastic agent bicalutamide [122,123,124,125]. These structures revealed that the CYP46A1 active site can accommodate compounds of different shapes, sizes, and polarity because of the conformational fit induced by these compounds.

Plasticity of the CYP46A1 active site. The enclosed volumes of the CYP46A1 active site, substrate-free and in complex with different compounds, are shown as semitransparent surfaces. Compounds inside the active site are shown as stick models. The heme group, which is the site of catalysis in the P450 enzymes, is in red
A remarkable finding from the screening of > 300 FDA-approved drugs was that in vitro CYP46A1 activity may not only be inhibited by different medications but also activated by agomelatine and mirtazapine (antidepressants), huperzine and galantamine (anti-AD medications), and acetaminophen and phenacetin (analgesics), as well as efavirenz (an anti-HIV drug) [121, 123]. This discovery gave impetus to animal experiments, which showed that voriconazole, the in vitro CYP46A1 inhibitor, also inhibits the P450 in mouse brain when given intraperitoneally and at a clinically relevant dose [116] (Table 1). Efavirenz was chosen as a CYP46A1 activator and was found to activate the enzyme in mouse brains at low concentrations but inhibit the P450 at higher concentrations [28]. Thus, it was proved that pharmacologic modulation of CYP46A1 activity is possible in vivo. To understand the mechanism of a dual efavirenz effect on CYP46A1 activity, studies with the purified P450 were carried out. These studies established that CYP46A1 has the allosteric site on the protein surface, which is away from the active site located inside the protein molecule [126]. Accordingly, at a low concentration, efavirenz was suggested to bind to the allosteric site and activate CYP46A1, whereas at high concentrations the drug likely binds to both allosteric and active sites and inhibits CYP46A1 because of the competition with cholesterol for the active site [28].
Efavirenz is usually given to HIV patients at the high dose of 600 mg/day, which is associated with the CNS toxicity and psychiatric events in 40–60% and 25–40% of patients, respectively (reviewed in Apostolova et al. [127]). Yet, the activating CYP46A1 dose in mice (0.1 mg/day/kg of body weight) equaled to about 7 mg/day if given to humans, which was a much lower dose than that given to HIV patients. Hence, efavirenz was tested for the effects on 5XFAD mice, an AD model, in which amyloid deposition begins at 2 months of age and behavioral deficits start to develop at 4 months of age [128]. Two efavirenz treatments were evaluated so far: the so-called 1st and 2nd treatment paradigms (Fig. 5, the results of the 2nd treatment paradigm will be published elsewhere). In both, efavirenz was given to mice in drinking water at the 0.1 mg/day/kg of body weight dose. In the 1st treatment paradigm, efavirenz was administered from 1 month of age (before amyloid plague appearance) to 9 months of age [29]; in the 2nd treatment paradigm, efavirenz dosing began at 3 months of age (after amyloid β deposition) and continued for 6 months. In both treatments, efavirenz activated CYP46A1, brain cholesterol turnover, and improved animal performance in the MWM test at 9 months of age. In addition, in the 2nd treatment paradigm, behavioral improvements were observed in the contextual fear conditioning tests. The principal difference between the two treatment paradigms was in efavirenz effects on the amyloid β load. In the 1st treatment paradigm, there were decreases in the amyloid β pathology as assessed by all the quantification methods: ELISA and stains of mouse cortex and hippocampus with Thioflavin S and 6E10, which mainly bind to the dense- and diffuse-core plaques, respectively. In contrast, in the 2nd treatment paradigm, there were no decreases in the amyloid β burden as assessed by ELISA and 6E10 stain but there were small reductions by 17–20% in the number and area of Thioflavin S-positive plaques in the cortex and hippocampus. Thus, in contrast to the 1st paradigm, which effectively decreased amyloid β load in the brain [29], the 2nd paradigm did not have such a pronounced effect on the amyloid β pathology. This difference is likely due to the lack or presence of amyloid plaques at the initiation of treatment and different treatment durations (6 or 8 months).

A comparative summary of the long-term efavirenz treatments of 5XFAD mice. Upward arrows, downward arrows, and left-right arrows indicate increase, decrease, and no change, respectively. The data are taken from Mast et al. 29 (the 1st treatment paradigm) and will be published elsewhere (the 2nd treatment paradigm)
An interesting observation provided by the 1st treatment paradigm was that cholesterol was depleted from mouse brains during the first 3 months of efavirenz treatment. This effect was linked to the young age of mice, whose brain and pool of brain cholesterol expand until the age of 3 months (reviewed in Dietschy and Turley [1]). Accordingly, when 1-month-old mice were put on efavirenz treatment in the 1st treatment paradigm, CYP46A1 activation and enhancement of the brain cholesterol turnover slowed down their cholesterol accumulation in the brain, and this effect was observed until the age of 3 months when animals became mature. Simultaneously with cholesterol depletion, young efavirenz-treated mice had impaired performance in the Y- and Morriz water maze tests, yet improved performance in the contextual fear conditioning test [29]. Therefore, changes in behavioral performance of young efavirenz-treated mice were attributed to cholesterol depletion from the brain during the first 3 months of efavirenz dosing. Nevertheless, despite transient behavioral impairments, the 1st and 2nd treatment paradigms clearly demonstrated that a long-term pharmacologic CYP46A1 activation could be beneficial for AD. These studies also showed that efavirenz effects are context-specific and served as a basis for the proof-of-concept clinical research study of efavirenz in patients with AD (ClinicalTrials.gov, NCT03706885). The mechanisms of efavirenz effect on behavioral improvements in the 1st (amyloid-decreasing) and 2nd (amyloid-unchanging) paradigms are currently under investigation and will be published elsewhere. If briefly, efavirenz treatment seems to target microglia and lipid rafts as well as significantly reduce protein phosphorylation. Other processes (plasmin-dependent amyloid clearance, inflammation, oxidative stress, cellular hypoxia, autophagy, ubiquitin-proteasome systems, and apoptosis) could be affected as well, in part via changes in the Ca2+-, small GTPase-, and catenin-signaling.
Besides in 5XFAD mice, efavirenz was tested in other experimental settings. Under the conditions of depression, the treatment of Swiss albino mice with the CYP46A1-activating efavirenz dose of 0.1 mg/kg body for 2 weeks decreased the immobility time in the forced swim test and increased sucrose intake as well as the brain serotonin levels. These results indicate a possible antidepressant efavirenz effect [113]. The CYP46A-inhibiting efavirenz dose of 15 mg/kg body weight was given with chow for 10 days to 8-month-old Tg2576 mice producing amyloid β. These mice had increases in the soluble amyloid β production and expression of β-secretase along with a reduction in microglial amyloid β phagocytosis as indicated by the cell culture studies [117]. Lastly, efficacy and toxicity screening in induced pluripotent stem cell–derived astrocytes and neurons showed that CYP46A1 activation by efavirenz reduces phosphorylated tau in early human AD neurons without affecting astrocyte viability [129]. Thus, evidence accumulates that efavirenz-induced CYP46A1 activation is generally beneficial, whereas enzyme inhibition could be deleterious in the context of AD.
CYP46A1 as a Target in Clinical Trials
Currently, CYP46A1 is investigated as a pharmacologic target in two clinical trials registered at ClinicalTrials.gov: NCT03706885 and NCT03650452. As of the time of this review writing, both trials are in the patient recruitment phase. In the NCT03706885 trial, CYP46A1 will be evaluated for activation by the 50 mg/day or 200 mg/day efavirenz dose that will be given to stable patients with mild cognitive impairment due to AD. This is a proof-of-concept, two-center, randomized, double-blind, placebo-controlled clinical research study with the anticipated enrollment of 36 participants. Efavirenz will be evaluated for safety and tolerability in the geriatric population as well as the effect on CYP46A1 activity in the brain as indicated by changes in the serum 24HC levels (the primary endpoints). The secondary endpoint is a unique stable isotope kinetics labeling study, in which some patients will be given deuterated water to precisely measure their CYP46A1 activation in the brain and brain cholesterol turnover. In addition, all participants will be genotyped for the APOE isoform status (ε2, ε3, or ε4) and presence of specific SNPs in CYP46A1 and CYP2B6 (the major efavirenz-metabolizing enzyme). A post hoc analysis will then be conducted to investigate whether these genetic factors affect a study participant’s response to efavirenz treatment (the tertiary endpoint). Neurocognitive assessments and the measurements of amyloid β, total tau, and phospho-tau in the patient CSF will be conducted as well but are not among the trial endpoints.
In the NCT03650452 trial, CYP46A1 will be evaluated for inhibition by the drug called TAK-935 (OV935) developed by the Takeda Pharmaceutical Company. This is a phase 2, multicenter, randomized, double-blind, placebo-controlled study with the anticipated enrollment of 126 participants. TAK-935 (OV935) will be given as an adjunctive therapy to pediatric patients with Dravet syndrome (also known as severe myoclonic epilepsy of infancy) or Lennox–Gastaut syndrome (a severe form of epilepsy that typically becomes apparent during infancy or early childhood). TAK-935 (OV935) will be assessed for the efficacy, safety, and tolerability as compared to placebo. The primary endpoint of this trial is a percent change from baseline in frequency of all seizures (convulsive and drop) per 28 days on TAK-935 (OV935) therapy as compared to placebo. The secondary endpoints are multiple and include the correlation of the TAK-935 concentration and plasma 24HC levels. Thus, clinical data will ultimately become available on the effects of CYP46A1 activation and inhibition in different pathologic conditions.
Conclusions
There have been a tremendous increase in our knowledge of CYP46A1 since the finding in 1996 that the flux of 24HC from the brain is important for cholesterol homeostasis in the CNS [20] and subsequent cloning of Cyp46a1 in 1999 [17]. Nevertheless, the significance of this enzyme for brain functions and potential as a therapeutic target for diseases of the brain are not yet fully understood and require further intensive investigations. These investigations should be conducted in vivo and with the appreciation that CYP46A1 activity can affect the brain cholesterol and 24HC levels as well as the rate of brain cholesterol turnover. In some conditions, these CYP46A1 effects will likely be uncoupled, thus enabling studies of the individual contributions of the CYP46A1 effects. We envision that the use of unbiased omics approaches on well-characterized samples of the whole brain, specific brain regions, and cell types will provide major advances in our understanding of the CYP46A1 role in health and disease conditions. Further development of genetic and pharmacologic tools that target CYP46A1 in specific brain regions or cell types will be of immense clinical and fundamental importance as well. Finally, there should be more clinical trials testing CYP46A1 as a therapeutic target for different brain diseases and conditions.
Abbreviations
- 24HC:
-
24-Hydroxycholesterol
- ACAT1:
-
Acyl-coenzyme A:cholesterol acyltransferase 1
- AD:
-
Alzheimer’s disease
- APOA1:
-
Apolipoprotein A1
- CSF:
-
Cerebrospinal fluid
- DHCR7:
-
7-Dehydrocholesterol reductase
- DHCR24:
-
∆24-Dehydrocholesterol reductase
- HD:
-
Huntington’s disease
- LCAT:
-
Lecithin:cholesterol acyltransferase
- LXRs:
-
Liver X receptors
- MWM:
-
Morris water maze
- NMDARs:
-
N-Methyl-d-aspartate receptors
- NPC1:
-
Niemann-Pick C1 protein
- NPC2:
-
Niemann-Pick C2 protein
- PD:
-
Parkinson’s disease
- SOAT:
-
Sterol O-acyltransferase
References
Dietschy JM, Turley SD (2004). Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 45: 1375–1397
Dietschy JM, Turley SD (2001). Cholesterol metabolism in the brain. Curr Opin Lipidol 12: 105–112
Dietschy JM (2009). Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem 390: 287–293
Pfrieger FW, Ungerer N (2011). Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 50: 357–371
Waterham HR, Koster J, Romeijn GJ, et al. (2001). Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet 69: 685–694
Waterham HR, Wijburg FA, Hennekam RC, et al. (1998). Smith-Lemli-Opitz syndrome is caused by mutations in the 7-dehydrocholesterol reductase gene. Am J Hum Genet 63: 329–338
Takamori S, Holt M, Stenius K, et al. (2006). Molecular anatomy of a trafficking organelle. Cell 127: 831–846
Pasinetti GM, Johnson SA, Oda T, et al. (1994). Clusterin (SGP-2): a multifunctional glycoprotein with regional expression in astrocytes and neurons of the adult rat brain. J Comp Neurol 339: 387–400
DeMattos RB, Brendza RP, Heuser JE, et al. (2001). Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE transgenic mice. Neurochem Int 39: 415–425
Elliott DA, Weickert CS, Garner B (2010). Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol 51: 555–573
Arenas F, Garcia-Ruiz C, Fernandez-Checa JC (2017). Intracellular Cholesterol Trafficking and Impact in Neurodegeneration. Front Mol Neurosci 10: 382
Bjorkhem I, Meaney S (2004). Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol 24: 806–815
Mak AC, Pullinger CR, Tang LF, et al. (2014). Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol 71: 1228–1236
Vance JE, Karten B (2014). Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J Lipid Res 55: 1609–1621
Andersson S, Davis DL, Dahlback H, et al. (1989). Cloning, structure, and expression of the mitochondrial cytochrome P-450 sterol 26-hydroxylase, a bile acid biosynthetic enzyme. J Biol Chem 264: 8222–8229
Stoffel-Wagner B (2003). Neurosteroid biosynthesis in the human brain and its clinical implications. Ann N Y Acad Sci 1007: 64–78
Lund EG, Guileyardo JM, Russell DW (1999). cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A 96: 7238–7243
Lund EG, Xie C, Kotti T, et al. (2003). Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem 278: 22980–22988
Bjorkhem I, Lutjohann D, Diczfalusy U, et al. (1998). Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res 39: 1594–1600
Lutjohann D, Breuer O, Ahlborg G, et al. (1996). Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A 93: 9799–9804
Meaney S, Bodin K, Diczfalusy U, et al. (2002). On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: critical importance of the position of the oxygen function. J Lipid Res 43: 2130–2135
Bretillon L, Diczfalusy U, Bjorkhem I, et al. (2007). Cholesterol-24S-hydroxylase (CYP46A1) is specifically expressed in neurons of the neural retina. Curr Eye Res 32: 361–366
Liao WL, Heo GY, Dodder NG, et al. (2011). Quantification of cholesterol-metabolizing P450s CYP27A1 and CYP46A1 in neural tissues reveals a lack of enzyme-product correlations in human retina but not human brain. J Proteome Res 10: 241–248
Brown J, 3rd, Theisler C, Silberman S, et al. (2004). Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem 279: 34674–34681
Ramirez DM, Andersson S, Russell DW (2008). Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol 507: 1676–1693
Russell DW, Halford RW, Ramirez DM, et al. (2009). Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem 78: 1017–1040
Maioli S, Bavner A, Ali Z, et al. (2013). Is it possible to improve memory function by upregulation of the cholesterol 24S-hydroxylase (CYP46A1) in the brain? PLoS One 8: e68534-e68534
Mast N, Li Y, Linger M, et al. (2014). Pharmacologic stimulation of cytochrome P450 46A1 and cerebral cholesterol turnover in mice. J Biol Chem 289: 3529–3538
Mast N, Saadane A, Valencia-Olvera A, et al. (2017). Cholesterol-metabolizing enzyme cytochrome P450 46A1 as a pharmacologic target for Alzheimer’s disease. Neuropharmacology 123: 465–476
Bretillon L, Lutjohann D, Stahle L, et al. (2000). Plasma levels of 24S-hydroxycholesterol reflect the balance between cerebral production and hepatic metabolism and are inversely related to body surface. J Lipid Res 41: 840–845
Zhang J, Liu Q (2015). Cholesterol metabolism and homeostasis in the brain. Protein Cell 6: 254–264
Bryleva EY, Rogers MA, Chang CC, et al. (2010). ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci U S A 107: 3081–3086
Chang TY, Chang CC, Cheng D (1997). Acyl-coenzyme A:cholesterol acyltransferase. Annu Rev Biochem 66: 613–638
Hirsch-Reinshagen V, Donkin J, Stukas S, et al. (2009). LCAT synthesized by primary astrocytes esterifies cholesterol on glia-derived lipoproteins. J Lipid Res 50: 885–893
Lutjohann D, Papassotiropoulos A, Bjorkhem I, et al. (2000). Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res 41: 195–198
Bretillon L, Siden A, Wahlund LO, et al. (2000). Plasma levels of 24S-hydroxycholesterol in patients with neurological diseases. Neurosci Lett 293: 87–90
Kolsch H, Heun R, Kerksiek A, et al. (2004). Altered levels of plasma 24S- and 27-hydroxycholesterol in demented patients. Neurosci Lett 368: 303–308
Heverin M, Bogdanovic N, Lutjohann D, et al. (2004). Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J Lipid Res 45: 186–193
Bogdanovic N, Bretillon L, Lund EG, et al. (2001). On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci Lett 314: 45–48
Jia F, Liu Z, Song N, et al. (2016). The association between CYP46A1 rs4900442 polymorphism and the risk of Alzheimer’s disease: A meta-analysis. Neurosci Lett 620: 83–87
Jin C, Zhang F, Zhu J, et al. (2013). Association of CYP46 gene polymorphism with sporadic Alzheimer’s disease in Chinese Han populations: a meta-analysis. Int J Neurosci 123: 226–232
Li M, Wang W, Li Y, et al. (2013). CYP46A1 intron-2T/C polymorphism and Alzheimer’s disease: an updated meta-analysis of 16 studies including 3,960 cases and 3,828 controls. Neurosci Lett 549: 18–23
Gibson Wood W, Eckert GP, Igbavboa U, et al. (2003). Amyloid beta-protein interactions with membranes and cholesterol: causes or casualties of Alzheimer’s disease. Biochim Biophys Acta 1610: 281–290
Martin MG, Pfrieger F, Dotti CG (2014). Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep 15: 1036–1052
Sparks DL (1997). Coronary artery disease, hypertension, ApoE, and cholesterol: a link to Alzheimer’s disease? Ann N Y Acad Sci 826: 128–146
Eckert GP, Cairns NJ, Maras A, et al. (2000). Cholesterol modulates the membrane-disordering effects of beta-amyloid peptides in the hippocampus: specific changes in Alzheimer’s disease. Dement Geriatr Cogn Disord 11: 181–186
Molander-Melin M, Blennow K, Bogdanovic N, et al. (2005). Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J Neurochem 92: 171–182
Gylys KH, Fein JA, Yang F, et al. (2007). Increased cholesterol in Abeta-positive nerve terminals from Alzheimer’s disease cortex. Neurobiol Aging 28: 8–17
Mori T, Paris D, Town T, et al. (2001). Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice. J Neuropathol Exp Neurol 60: 778–785
Roher AE, Weiss N, Kokjohn TA, et al. (2002). Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry 41: 11080–11090
Corder EH, Saunders AM, Strittmatter WJ, et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: 921–923
Mayeux R, Stern Y, Ottman R, et al. (1993). The apolipoprotein epsilon 4 allele in patients with Alzheimer’s disease. Ann Neurol 34: 752–754
Corder EH, Saunders AM, Risch NJ, et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 7: 180–184
Lambert JC, Heath S, Even G, et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41: 1094–1099
Mahley RW, Rall SC, Jr. (2000). Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 1: 507–537
Mahley RW (2016). Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler Thromb Vasc Biol 36: 1305–1315
Bu G (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10: 333–344
Kremer B, Goldberg P, Andrew SE, et al. (1994). A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N Engl J Med 330: 1401–1406
Reiner A, Albin RL, Anderson KD, et al. (1988). Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A 85: 5733–5737
Leoni V, Mariotti C, Tabrizi SJ, et al. (2008). Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain 131: 2851–2859
Leoni V, Mariotti C, Nanetti L, et al. (2011). Whole body cholesterol metabolism is impaired in Huntington’s disease. Neurosci Lett 494: 245–249
Leoni V, Long JD, Mills JA, et al. (2013). Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol Dis 55: 37–43
Boussicault L, Alves S, Lamaziere A, et al. (2016). CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 139: 953–970
Kreilaus F, Spiro AS, McLean CA, et al. (2016). Evidence for altered cholesterol metabolism in Huntington’s disease post mortem brain tissue. Neuropathol Appl Neurobiol 42: 535–546
del Toro D, Xifro X, Pol A, et al. (2010). Altered cholesterol homeostasis contributes to enhanced excitotoxicity in Huntington’s disease. J Neurochem 115: 153–167
Valenza M, Rigamonti D, Goffredo D, et al. (2005). Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J Neurosci 25: 9932–9939
Lee VM, Trojanowski JQ (2006). Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52: 33–38
Bjorkhem I, Lovgren-Sandblom A, Leoni V, et al. (2013). Oxysterols and Parkinson’s disease: evidence that levels of 24S-hydroxycholesterol in cerebrospinal fluid correlates with the duration of the disease. Neurosci Lett 555: 102–105
Huang X, Sterling NW, Du G, et al. (2019). Brain cholesterol metabolism and Parkinson’s disease. Mov Disord 34: 386-395
Di Natale C, Monaco A, Pedone C, et al. (2018). The level of 24-hydroxycholesteryl esters decreases in plasma of patients with Parkinson’s disease. Neurosci Lett 672: 108–112
Cheng D, Jenner AM, Shui G, et al. (2011). Lipid pathway alterations in Parkinson’s disease primary visual cortex. PLoS One 6: e17299
Wolozin B, Wang SW, Li NC, et al. (2007). Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med 5: 20
Rozani V, Gurevich T, Giladi N, et al. (2018). Higher serum cholesterol and decreased Parkinson’s disease risk: A statin-free cohort study. Movement Disorders 33: 1298–1305
Wahner AD, Bronstein JM, Bordelon YM, et al. (2008). Statin use and the risk of Parkinson disease. Neurology 70: 1418–1422
Undela K, Gudala K, Malla S, et al. (2013). Statin use and risk of Parkinson’s disease: a meta-analysis of observational studies. J Neurol 260: 158–165
de Lau LM, Koudstaal PJ, Hofman A, et al. (2006). Serum cholesterol levels and the risk of Parkinson’s disease. Am J Epidemiol 164: 998–1002
Simon KC, Chen H, Schwarzschild M, et al. (2007). Hypertension, hypercholesterolemia, diabetes, and risk of Parkinson disease. Neurology 69: 1688–1695
Huang X, Abbott RD, Petrovitch H, et al. (2008). Low LDL cholesterol and increased risk of Parkinson’s disease: prospective results from Honolulu-Asia Aging Study. Mov Disord 23: 1013–1018
Kalaany NY, Mangelsdorf DJ (2006). LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol 68: 159–191
Glass CK, Ogawa S (2006). Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol 6: 44–55
Calkin AC, Tontonoz P (2012). Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol 13: 213–224
Courtney R, Landreth GE (2016). LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol Metab 27: 404–414
Shafaati M, Olin M, Bavner A, et al. (2011). Enhanced production of 24S-hydroxycholesterol is not sufficient to drive liver X receptor target genes in vivo. J Intern Med 270: 377–387
Prufer K, Boudreaux J (2007). Nuclear localization of liver X receptor alpha and beta is differentially regulated. J Cell Biochem 100: 69–85
Ishikawa T, Yuhanna IS, Umetani J, et al. (2013). LXRbeta/estrogen receptor-alpha signaling in lipid rafts preserves endothelial integrity. J Clin Invest 123: 3488–3497
Collingridge GL, Volianskis A, Bannister N, et al. (2013). The NMDA receptor as a target for cognitive enhancement. Neuropharmacology 64: 13–26
Paul SM, Doherty JJ, Robichaud AJ, et al. (2013). The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J Neurosci 33: 17290–17300
Sun MY, Izumi Y, Benz A, et al. (2016). Endogenous 24S-hydroxycholesterol modulates NMDAR-mediated function in hippocampal slices. J Neurophysiol 115: 1263–1272
Wei X, Nishi T, Kondou S, et al. (2018). Preferential enhancement of GluN2B-containing native NMDA receptors by the endogenous modulator 24S-hydroxycholesterol in hippocampal neurons. Neuropharmacology 148: 11–20
Miller OH, Yang L, Wang CC, et al. (2014). GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife 3: e03581
Xia P, Chen HS, Zhang D, et al. (2010). Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci 30: 11246–11250
Coyle JT (2006). Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol 26: 365–384
Sun MY, Linsenbardt AJ, Emnett CM, et al. (2016). 24(S)-Hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist 22: 132–144
Sodero AO, Vriens J, Ghosh D, et al. (2012). Cholesterol loss during glutamate-mediated excitotoxicity. EMBO J 31: 1764–1773
Mast N, Anderson KW, Johnson KM, et al. (2017). In vitro cytochrome P450 46A1 (CYP46A1) activation by neuroactive compounds. J Biol Chem 292: 12934-12946
Sezgin E, Levental I, Mayor S, et al. (2017). The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol 18: 361–374
Grouleff J, Irudayam SJ, Skeby KK, et al. (2015). The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. Biochim Biophys Acta 1848: 1783–1795
Mauch DH, Nagler K, Schumacher S, et al. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294: 1354–1357
Koudinov AR, Koudinova NV (2001). Essential role for cholesterol in synaptic plasticity and neuronal degeneration. Faseb J 15: 1858–1860
Frank C, Rufini S, Tancredi V, et al. (2008). Cholesterol depletion inhibits synaptic transmission and synaptic plasticity in rat hippocampus. Exp Neurol 212: 407–414
Sebastiao AM, Colino-Oliveira M, Assaife-Lopes N, et al. (2013). Lipid rafts, synaptic transmission and plasticity: impact in age-related neurodegenerative diseases. Neuropharmacology 64: 97–107
Egawa J, Pearn ML, Lemkuil BP, et al. (2016). Membrane lipid rafts and neurobiology: age-related changes in membrane lipids and loss of neuronal function. J Physiol 594: 4565–4579
Mast N, Lin JB, Anderson KW, et al. (2017). Transcriptional and post-translational changes in the brain of mice deficient in cholesterol removal mediated by cytochrome P450 46A1 (CYP46A1). PLoS One 12: e0187168
Kotti TJ, Ramirez DM, Pfeiffer BE, et al. (2006). Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci U S A 103: 3869–3874
Ohyama Y, Meaney S, Heverin M, et al. (2006). Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J Biol Chem 281: 3810–3820
Shafaati M, O’Driscoll R, Bjorkhem I, et al. (2009). Transcriptional regulation of cholesterol 24-hydroxylase by histone deacetylase inhibitors. Biochem Biophys Res Commun 378: 689–694
Milagre I, Nunes MJ, Moutinho M, et al. (2010). Chromatin-modifying agents increase transcription of CYP46A1, a key player in brain cholesterol elimination. J Alzheimers Dis 22: 1209–1221
Nunes MJ, Milagre I, Schnekenburger M, et al. (2010). Sp proteins play a critical role in histone deacetylase inhibitor-mediated derepression of CYP46A1 gene transcription. J Neurochem 113: 418–431
Milagre I, Nunes MJ, Gama MJ, et al. (2008). Transcriptional regulation of the human CYP46A1 brain-specific expression by Sp transcription factors. J Neurochem 106: 835–449
Milagre I, Nunes MJ, Castro-Caldas M, et al. (2012). Neuronal differentiation alters the ratio of Sp transcription factors recruited to the CYP46A1 promoter. J Neurochem 120: 220–229
Burlot MA, Braudeau J, Michaelsen-Preusse K, et al. (2015). Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum Mol Genet 24: 5965–5976
Hudry E, Van Dam D, Kulik W, et al. (2010). Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol Ther 18: 44–53
Patel TK, Patel VB, Rana DG (2017). Possible anti-depressant effect of efavirenz and pro-depressive-like effect of voriconazole in specified doses in various experimental models of depression in mice. Pharmacol Rep 69: 1082–1087
Djelti F, Braudeau J, Hudry E, et al. (2015). CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 138: 2383–2398
Chali F, Djelti F, Eugene E, et al. (2015). Inhibiting cholesterol degradation induces neuronal sclerosis and epileptic activity in mouse hippocampus. Eur J Neurosci 41: 1345–1355
Shafaati M, Mast N, Beck O, et al. (2010). The antifungal drug voriconazole is an efficient inhibitor of brain cholesterol 24S-hydroxylase (CYP46A1) in vitro and in vivo. J Lipid Res 51: 318–323
Brown LA, Jin J, Ferrell D, et al. (2014). Efavirenz promotes beta-secretase expression and increased Abeta1-40,42 via oxidative stress and reduced microglial phagocytosis: implications for HIV associated neurocognitive disorders (HAND). PLoS One 9: e95500
Kotti T, Head DD, McKenna CE, et al. (2008). Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proc Natl Acad Sci U S A 105: 11394–11399
Halford RW, Russell DW (2009). Reduction of cholesterol synthesis in the mouse brain does not affect amyloid formation in Alzheimer’s disease, but does extend lifespan. Proc Natl Acad Sci U S A 106: 3502–3506
Mast N, Norcross R, Andersson U, et al. (2003). Broad substrate specificity of human cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry 42: 14284–14292
Mast N, White MA, Bjorkhem I, et al. (2008). Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc Natl Acad Sci U S A 105: 9546–9551
Mast N, Charvet C, Pikuleva IA, et al. (2010). Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem 285: 31783–31795
Mast N, Linger M, Clark M, et al. (2012). In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol Pharmacol 82: 824–834
Mast N, Zheng W, Stout CD, et al. (2013). Binding of a cyano- and fluoro-containing drug bicalutamide to cytochrome P450 46A1: unusual features and spectral response. J Biol Chem 288: 4613–4624
Mast N, Zheng W, Stout CD, et al. (2013). Antifungal azoles: structural insights into undesired tight binding to cholesterol-metabolizing CYP46A1. Mol Pharmacol 84: 86–94
Anderson KW, Mast N, Hudgens JW, et al. (2016). Mapping of the allosteric site in cholesterol hydroxylase CYP46A1 for efavirenz, a drug that stimulates enzyme activity. J Biol Chem 291: 11876–11886
Apostolova N, Funes HA, Blas-Garcia A, et al. (2015). Efavirenz and the CNS: what we already know and questions that need to be answered. J Antimicrob Chemother 70: 2693-2708
Oakley H, Cole SL, Logan S, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26: 10129–10140
van der Kant R, Langness VF, Herrera CM, et al. (2019). Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-beta in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 24: 363–375 e369
Acknowledgments
We thank Dr. Natalia Mast for help with the figure preparation. Some of the studies described in this review were supported in part by the National Institute of General Medical Sciences Grant GM062882 and the Alzheimer’s Drug Discovery Foundation Grant 20160601 (Irina A. Pikuleva).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 498 kb)
Rights and permissions
About this article

Cite this article
Petrov, A.M., Pikuleva, I.A. Cholesterol 24-Hydroxylation by CYP46A1: Benefits of Modulation for Brain Diseases. Neurotherapeutics 16, 635–648 (2019). https://doi.org/10.1007/s13311-019-00731-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00731-6