
Abstract
The immune response to acute cerebral ischemia is a major factor in stroke pathobiology and outcome. While the immune response starts locally in occluded and hypoperfused vessels and the ischemic brain parenchyma, inflammatory mediators generated in situ propagate through the organism as a whole. This “spillover” leads to a systemic inflammatory response first, followed by immunosuppression aimed at dampening the potentially harmful proinflammatory milieu. In this overview we will outline the inflammatory cascade from its starting point in the vasculature of the ischemic brain to the systemic immune response elicited by brain ischemia. Potential immunomodulatory therapeutic approaches, including preconditioning and immune cell therapy will also be discussed.
Similar content being viewed by others


Introduction
Despite recent therapeutic advances, ischemic stroke remains a vexing problem with immense socioeconomic impact worldwide [1]. Acute ischemic stroke is the most common stroke type, accounting for > 80 % of all stroke [2]. Therapeutic approaches have been primarily directed to preserve neurons in the ischemic territory and both Food and Drug Administration-approved treatments currently available in clinics—intra-arterial recombinant tissue plasminogen activator and endovascular intervention—aim at fast recanalization to restore oxygen and nutrient supplies to the affected area [3]. In the core of the ischemic territory, clinically defined as the region with regional cerebral blood flow < 20 %, acute neuronal death occurs rapidly, within minutes to hours, after the occlusion, owing to an energy deficit resulting in intracellular ionic imbalance, mitochondrial failure, and activation of intracellular proteases, lipases, and ribonucleases leading to fast breakdown of cellular structural elements and to loss of cell integrity. However, outside the ischemic core the brain tissue is still partially perfused although at reduced rate. This region, referred to as ischemic penumbra, is often defined by a reduced perfusion rate that is, however, greater than the one observed in the ischemic core [4]. While penumbral neurons are functionally compromised, they are salvageable if blood flow is restored [2]. However, even if blood flow is restored, neurons in the penumbra face major challenges to their survival, such as excitotoxicity and inflammation. Excitotoxicity stems from the uncontrolled release of the neurotransmitter glutamate from depolarizing or dying neurons. Glutamate-mediated activation of N-methyl-D-aspartate and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, potentiated by prolonged activation of transient receptor potential melastatin ion channels, leads to uncontrolled extracellular calcium influx and dysregulation of intracellular calcium homeostasis that results in generation of reactive oxygen (ROS) and nitrogen species, mitochondrial dysfunction, activation of the apoptotic cascade, and poly (adenosine diphosphate-ribose) polymerase activation [2].
Inflammation, initiated by stagnant blood flow, activation of intravascular leukocytes, and release of proinflammatory mediators from the ischemic endothelium and brain parenchyma, has the potential to increase tissue injury. As reviewed in the next sections, while many aspects of inflammation are beneficial and aimed at restoring tissue homeostasis, collateral damage by the acute inflammatory response contributes to the ischemic damage.
Intravascular Initiation of the Inflammatory Response
Although many aspects of postischemic inflammation, that is, the response of the immune system to disruption of tissue homeostasis, manifest themselves days and weeks after the event, the inflammatory cascade is activated immediately after vessel occlusion has occurred [5, 6]. Stagnant blood flow and altered rheology induce shear stress on the vascular endothelium and platelets. This results in the deployment of the adhesion molecule P-selectin to the cell surface. P-selectin is stored in Weibel–Palade bodies in the endothelium and α-granules in platelets and can be released within minutes after activation. Selectins are crucial in slowing down circulating leukocytes by attracting them to the endothelial surface by interacting with P-selectin glycoprotein ligand-1 expressed on most leukocytes. Platelet P-selectin can bind also to leukocytes and act as a bridging molecule that facilitates cluster formation of leukocytes causing intravascular clogging that further contributes to ischemic damage [6]. Other adhesion molecules are rapidly induced at the transcriptional level after activation of endothelial pattern recognition and cytokine receptors. Among them, E-selectin, intercellular adhesion molecule 1 and vascular cell adhesion molecule 1 play an important role in orchestrating blood leukocyte recruitment, adhesion and transmigration [7].
Activation of the coagulation cascades further generates inflammatory cues [8]. Thrombin is a chemotaxin for monocytes and neutrophils and induces expression of adhesion molecules on endothelial cells through activation of protease-activated receptors and nucleaer factor kappa B [9]. Thrombin also directly activates both C3 and C5 components of the complement system and can disrupt endothelial barrier function [10]. Furthermore, loss of endothelial thrombomodulin expression, which, in conjunction with activated protein C, endothelial protein C receptor, and protein S, forms a strong anticoagulant and anti-inflammatory complex, enhances inflammation, and leads to monocytes and complement activation.
The complement system, the humoral branch of innate immunity, has been consistently implicated in the pathobiology of stroke and its activation is associated with unfavorable outcome [11, 12]. Bioactive products of the complement cascade consist of opsonins (iC3b, C3dg, C3d), anaphylatoxins (C3a, C5a), and a terminal cytolytic membrane attack complex. Activation occurs by 1 of 3 pathways, referred to as the classical, alternative, and lectin pathways. All pathways converge at the activation of the complement protein C3. In the setting of cerebral ischemia, the complement system might be activated intravascularly by proteases of the coagulation cascade, at the level of C3 and C5 cleavage, thus bypassing classical, alternative, and lectin pathways. However, activation of the alternative and lectin pathways might also contribute to ischemic brain injury. While intravascularly generated active complement proteins might gain access to the brain parenchyma through a compromised blood–brain barrier (BBB), there is also evidence of increased complement synthesis by microglia [13]. The mechanisms of complement-mediated neurotoxicity in brain ischemia are not fully understood, but clear evidence for a major role of membrane attack complex is missing. It is rather believed that anaphylatoxins (C3a, C5a) act on complement receptors found on most immune cells of myeloid origin to increase ROS production, secretion of proinflammatory cytokines, and degranulation. The C3a receptor has been most consistently implicated in stroke pathobiology and C3–/– mice or treatment with C3a receptor antagonist has been shown to reduce ischemic brain injury and to improve functional outcome [14]. There is evidence that the lectin pathway might contribute to ischemic brain injury and mice deficient in mannose binding-lectin, the major activator of this cascade, were protected after transient middle cerebral artery (MCA) occlusion [15]. Moreover, mannose binding-lectin deficiency in humans was correlated with better stroke outcome [15, 16].
While this intravascular inflammation sets the stage for BBB breakdown and leukocyte invasion of the ischemic tissue, inflammatory processes are also initiated in the brain parenchyma. One of the earliest events might be the release of danger-/damage-associated molecular patterns (DAMP) from injured and dying neurons. Such DAMPs, including purines (ATP, UTP, and their catabolites), high mobility group binding protein 1 (HMGB1), heat shock proteins, peroxiredoxins, and mitochondrial-derived N-formyl peptides, activate pattern recognition receptors on microglia, the brain resident immune cells, and astrocytes [7, 17–19]. Microglia respond to the altered environment within hours of onset of ischemia, as evidenced by altered morphology and increased contact with neurons that showed signs of excitotoxic calcium overload [20]. PPR activation on microglia leads to inflammasome mediated interleukin (IL)-1β release, as well as tumor necrosis factor (TNF) production, which feed back into the inflammatory cascade by inducing cytokine and chemokine production in endothelial cells and astrocytes.
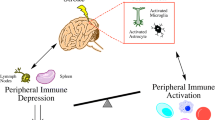
The Peripheral Immune Response to Stroke
DAMPs and cytokines expressed during this early phase of ischemic injury can gain access to the systemic circulation through the disrupted BBB or the cerebrospinal fluid (CSF) drainage system that includes venous and lymphatic outflows. It has been shown that HMGB-1 is increased in patients with acute ischemic stroke and that blocking the HMGB-1/RAGE axis provides better outcome after transient MCA occlusion in mice [21, 22]. Once in circulation DAMPs and cytokines induce a response of the immune system in primary and secondary lymphoid organs, resulting in a systemic inflammatory response syndrome. In experimental stroke, systemic inflammatory response syndrome is characterized by elevated serum cytokine levels [IL-6, interferon-γ, chemokine (C-X-C motif) ligand 1] and increased production of inflammatory mediators in circulating and splenic immune cells [TNF, IL-6, IL-2, C-C motif chemokine ligand 2, and chemokine (C-X-C motif) ligand 2] within hours of ischemia [23, 24]. The response is generally transient and most parameters return to baseline levels 24 h after stroke. Comparable changes can be observed in human stroke patients. TNF and IL-6 are increased in patient at stroke onset (<24 h) [25], and IL-6 serum levels are positively correlated with stroke severity and unfavorable outcome [26].
However, this early activation of the immune system is superseded by a state of systemic immunodepression that predisposes to poststroke infections [27, 28]. Accordingly, complication from pulmonary or urinary tract infections have been observed in ~20 % of patients with stroke [29]. Early studies on the immune status of patients with stroke found prolonged peripheral lymphopenia and reduced T-cell responsiveness [30]. Ischemic brain injury leads to sustained decrease in blood and splenic B, T, and natural killer (NK) cells in mice undergoing transient MCA occlusion [28, 31, 32]. These changes were observed 12 h after ischemia and persisted for several weeks. Importantly, the immunosuppression resulted in increased susceptibility to nosocomial bacterial infection and to higher mortality rate [28]. The decrease in lymphocytes was correlated with increased splenocyte apoptosis, spleen atrophy, and regulatory T cell (Treg) expansion [28, 31]. The sympathetic nervous system is fundamentally involved in this response. Treatment of animals with the β-adrenergic receptor antagonist propranolol, or chemical ablation of sympathetic noradrenergic terminals by 6-hydroxydopamine, were sufficient to lower bacteremia and bacterial colonization of the lungs, and increased significantly survival rates along with preservation of splenic and blood lymphocyte populations [28]. Interestingly, the glucocorticoid receptor antagonist RU-486 reversed the lymphopenia in the blood, but not in the spleen, and did not affect bacterial colonization or survival rates after stroke. Whether the splenic response to cerebral ischemia is induced by similar mechanisms in all stroke models and in humans remains to be determined. Studies in rats concluded that the loss of splenic lymphocytes was not due to increased apoptosis, but to activation of α-adrenergic receptors on trabecular and capsular smooth muscle cells that leads to spleen contraction and expels immune cells into circulation [33]. The study found that although spleen size was decreased 1 day after stroke, spleen volume was restored 3 days thereafter, arguing against long-lasting effects of cerebral ischemia on spleen physiology. As in rats, splenic size loss in humans is transient and there is a tendency of increased splenic volume 4 days after stroke [34]. The immunosuppressive effects of ischemic brain injury are not limited to the spleen. In the bone marrow, tyrosine hydroxylase and norepinephrine levels increase 1 day after transient MCA occlusion in mice [35]. This triggers a response in mesenchymal stromal cells, probably through activation of β3-adrenergic receptors, resulting in reduction of homeostatic and cell retention factors such as IL-7, C-X-C motif chemokine 12 (stromal cell-derived factor 1), vascular cell adhesion protein 1, stem cell factor, and angiopoietin-1. Downregulation of these factors increases hematopoietic stem cell proliferation. This proliferative response, however, does not involve all arms of blood cell lineages equally and hematopoietic system becomes skewed towards the myeloid lineage [35]. This switch is accomplished by increased expression of transcription factors associated with myeloid lineage progression such as the ETS-domain transcription factor PU.1 and CCAAT/enhancer-binding protein. Collectively, the data suggest that the injured brain exerts a strong influence on the peripheral immune system by regulating development and homeostasis of splenic and bone marrow immune cell populations and that increased sympathetic output after stroke is the main efferent branch responsible for these effects.
A third phase of the peripheral immune response to stroke involves sensitization to central nervous system (CNS)-derived neoantigens [5, 36, 37]. Disruption of the BBB and brain–CSF barrier during acute stroke releases novel CNS antigens that are normally sequestered in the brain and exposes them to the systemic immune system. There is an ever increasing list of CNS-derived peptides that can induce a peripheral T-cell response after stroke including, but not limited to, peptides derived from myelin basic protein, neuron-specific enolase, proteolipid protein, N-methyl-D-aspartate receptor 2A, and microtubule-associated protein [37]. Meningeal lymphatic vessels, that run along the venous sinus and terminate in the deep cervical lymph nodes [38], are likely to be involved in the cranial export of antigenic macromolecules and antigen-presenting cells after ischemic brain injury. Evidence for antigen-specific T-cell reactivity has been found in animal models of stroke. As early as 4 days after transient focal ischemia peptide-reactive B and T cells can be found in cervical lymph nodes and spleen in mice [39]. Among T cells, the response was similar in memory (CD4+) and effector (CD8+) T cells. In human stroke, antigen-presenting cells loaded with CNS-derived peptides have been found in T-cell zones of cervical lymph nodes and tonsils [40]. Association studies of this small patient cohort indicated that increased reactivity to neuronal-derived antigens was correlated with smaller infarct size and better long-term outcome, whereas greater reactivity to MBP was correlated with larger infarcts and worse outcome. This dichotomy raises the intriguing possibility that the adaptive immune response to ischemic brain injury can be skewed towards damaging [T helper cell (Th)1/Th17] or protective (Th2) phenotypes. This interpretation is supported by studies in rodent stroke models indicating a beneficial effect of a Th2 immune response on stroke outcome [41, 42]. Future studies will have to address whether a tolerogenic immune response is linked to favorable stroke outcome in humans.
Innate Immune Cells in Stroke
Microglia, Monocyte-Derived Macrophages, and Dendritic Cells
Microglia and monocyte-derived macrophages (MMΦ) seem to have largely protective functions in ischemic brain injury. Eliminating proliferating microglia/macrophages by ganciclovir treatment in mice that express herpes simplex virus-1 thymidine kinase in the myeloid lineage increased ischemic brain injury and reduced expression of the neurotrophic cytokine insulin-like growth factor 1 [43]. MMΦ develop from monocytes after infiltration into the brain parenchyma. Blood monocytes exist as 2 functionally distinct subpopulations (inflammatory and patrolling) best characterized by their expression of C-C chemokine receptor 2 and C-X3-C chemokine receptor 1, both of which are important for monocyte entry into the ischemic brain [44–46], and the abundance of inflammatory monocytes in the circulation has been linked to unfavorable clinical stroke outcome [47, 48]. Initially, infiltrating monocytes are of the “inflammatory” subtype while “patrolling” monocytes are prevalent at later time points [49]. This study also showed that wild-type mice transplanted with C-C chemokine receptor 2–/– bone marrow exhibited reduced monocyte infiltration after transient MCA occlusion but had higher hemorrhagic transformation rates hinting at a vasculoprotective role of MMΦ. Blocking “inflammatory” monocyte recruitment also abolished long-term behavioral recovery and increased proinflammatory cytokine expression in mice undergoing transient MCA occlusion [50]. In addition, depending on molecular cues encountered in the postischemic brain parenchyma, MMΦ can undergo classical (M1, proinflammatory) or alternative (M2, anti-inflammatory in most settings) activation, thus contributing to the resolution of inflammation [51, 52]. Phagocytosis, which is mainly seen in microglia and not MMΦ and is tied to MER proto-oncogene tyrosine kinase, milk fat globule–epidermal growth factor 8 protein, and triggering receptor expressed on myeloid cell 2 expression [53, 54], can contribute to delayed neuronal cell death after ischemia [54, 55]. A subset of microglia and infiltrating macrophages express the dendritic cell (DC) marker CD11c and major histocompatibility complex class II molecules [56]. Using chimeric animals that express yellow fluorescent protein under the control of the CD11c promoter in bone marrow-derived cells, the same study showed that DCs originate from brain-resident myeloid cells early after ischemia, while peripheral immune cells contribute the majority of CD11c+/ yellow fluorescent protein+ cells 3 days after stroke [56]. Although brain DCs have the capacity to present antigen and induce T-cell proliferation in vitro [57], it remains to be shown whether these cells engage in antigen presentation after brain ischemia, as predicted by some studies [58].
Neutrophils
Although intravascular adhesion of neutrophils is a relatively early postischemic event, parenchymal accumulation is generally observed later. Nevertheless, neutrophils are among the first hematogenous immune cells found in the brain after experimental stroke peaking at 48 to 72 h in most models and declining rapidly afterwards. Although it is not clear whether they enter the brain parenchyma under all circumstances [59], there is evidence that neutrophils contribute to postischemic inflammation by limiting tissue perfusion due to intravascular clogging [60, 61], destabilizing the BBB by releasing matrix metalloproteinases [62, 63], and by generating ROS and reactive nitrogen species [64, 65]. Recent data indicate that postischemic CSF3 production by cerebral endothelial cells is critical for activation of neutrophil neurotoxicity, a process requiring the scavenger receptor CD36 in cerebral endothelial cells [66]. However, a cause-and-effect relationship between the extent of neutrophil trafficking and the severity of ischemic damage has not been firmly established [67]. Attesting to the complex role of neutrophils in cerebral ischemic injury, a recent study postulates a protective role of neutrophils, that have undergone N2 polarization as characterized by Ym1 (chitinase 3-like 3, Chi3l3) expression, in stroke pathology [68].
Mast Cells
Mast cells are brain-resident immune cells located in the perivascular space surrounding brain parenchymal vessels and in the dura mater of the meninges, which is rich in peptidases and vasoactive molecules. Mast cells are activated early after cerebral ischemia and contribute to the BBB breakdown and brain edema by releasing gelatinase [69, 70].
Innate Lymphocytes
T cells are detrimental in the early phase of ischemia and lymphocyte-deficient mice are protected in models of focal ischemia [71, 72]. The mechanism does not involve classical antigen-mediated T-cell activation and the cytotoxic activity might be tied to innate T-cell functions [72]. Accordingly, IL-17-secreting γδT cells, which do not undergo classical antigen-dependent T-cell activation, have been shown to contribute to ischemic injury [73–75]. NK cells also contribute to ischemic brain injury [76]. Expression of C-X3-C chemokine receptor 1 on NK cells was required for the detrimental effect that was independent of T and B cells, but was dependent on perforin expression, pointing at a direct cytolytic activity of NK cells towards ischemic neurons [76].
Adaptive Immune Cells in Stroke
T Cells
While effector T lymphocytes may contribute to focal ischemic injury [71, 77], Tregs could have a protective effect by downregulating postischemic inflammation. Tregs appear in the ischemic tissue after the acute phase and confer neuroprotection by IL-10 secretion, an effect that might be antigen independent [78–81]. However, the presence of intravascular Treg during the reperfusion phase after transient MCA occlusion might contribute to ischemic brain injury by causing vascular dysfunction and thrombosis [82]. There is less evidence for a role of “classical” antigen-dependent T-cell response in determining stroke outcome. While T cells respond to CNS neoantigens after stroke (see above), the contribution of antigen specific T cells in the long-term outcome after stroke remains a matter of debate [83]. Future studies will have to address whether clonal expansion of CNS-reactive T cells directly affects injury development during the chronic phase of stroke or whether these cells exhibit a peripheral immunomodulatory function.
B Cells
B cells have been shown to exhibit both detrimental and protective properties. In mouse models, B cells are involved in the chronic inflammatory response in the ischemic territory where organized lymphoid tissue similar to B-cell follicles has been observed [84]. The B cells undergo isotype switching, express plasma cell markers, and secrete immunoglobulins to impair long-term functional recovery after stroke. Concordantly, oligoclonal immunoglobulins can be found in the CSF of some patients with stroke, suggesting a B-cell response to stroke within the CNS [85, 86]. Similarly to Tregs, regulatory B cells confer neuroprotection through an IL-10-dependent mechanism, but do not enter the ischemic brain [87–89].
The Microbiota–Gut–Brain Axis in Stroke
One of the largest immune cell compartments in the body is the gastrointestinal tract (GI). This is, in part, because the intestine is home to trillions of commensal bacteria that expose the GI immune system continuously to bacterial antigens. There are 4 distinct immune-cell compartments in the GI. The Peyer’s patches (PP), immune cells of the lamina propria (LP), intraepithelial lymphocytes (IEL), and the mesenteric lymph nodes. The PP are structured as secondary lymphoid organs with well-separated T- and B-cell areas. The LP contains most components of the immune system, with large numbers of B cells, T cells of both the CD4 and CD8 subsets, macrophages, and DC. IELs compromise 10 % to 15 % of the normal epithelium and are predominantly CD8+ T and γδT cells.
Commensal gut bacteria are in direct contact with the intestinal epithelium and intestinal immune cells. Epithelial cells express an array of DAMP receptors and communicate bacterial signals through the release of cytokines to the immune cells located in the PP and LP. Moreover, specialized epithelial cells (M cells) can uptake and transport antigens to the submucosa. In addition, IELs and submucosal myeloid cells, located close to the luminal surface, can directly sense the presence of bacteria and their metabolites. Gut microbiota provide diverse signals for tuning the host immune system toward either effector or regulatory phenotype, and is thus critical for peripheral immune education and homeostasis [90, 91]. These interactions are vital for immune system development, as lack of gut microbiota in germ-free animals prevents the full development of the intestinal and peripheral immune system [92–94]. Recent studies have begun to elucidate the role of intestinal microbiota and GI immune cells in stroke. In a model of antibiotic-induced microbial dysbiosis, Benakis et al. [75] showed that treatment of mice with amoxicillin increased Tregs and suppressed IL17+γδT in the small intestine, which correlated with smaller infarcts and better behavioral outcome after transient MCA occlusion, an effect that could be transferred by fecal matter transplants [75]. Using photoconvertible reporter mice the same study showed trafficking of intestinal immune cells to the meninges after stroke. However, administration of an antibiotic cocktail that depletes the intestine of all cultivatable bacteria did not affect infarct volume at day 1 after MCA occlusion, but resulted in lower survival rates [95]. Stroke itself also has a profound effect on the composition of intestinal bacteria. It has been shown that increased sympathetic activity after MCA occlusion decreases gut motility and altered caecal mucoprotein secretion and microbial composition 3 days after stroke [96]. Similarly, Singh et al. [97] found that stroke decreased intestinal motility and induced intestinal dysbiosis, resulting in decreased microbial diversity. Importantly, transplantation of fecal matter from stroked mice into germ-free mice undergoing distal MCA occlusion 3 days after transplantation resulted in increased infarct size, higher IL-17 and interferon (IFN)-γ expression in the ischemic hemisphere, and increased Th1 and Th17 cells in the PP. Together these studies point to a bidirectional interaction of microbiota, the intestinal immune system, and the ischemic brain, where brain injury alters intestinal microbiota, which, in turn, affects stroke outcome by modulating post ischemic inflammatory responses.
Immunomodulation as a Therapeutic Approach: Preconditioning and Immune-Cell Therapies
Ischemic brain cells produce a large array of mediators that may participate in progression of ischemic brain injury. Surprisingly, these inflammatory mediators, under certain conditions, can induce tolerance to cerebral ischemia.
The earliest observations of tolerance were in the heart and involved preconditioning with brief sublethal ischemia [98]. Preconditioning is the phenomenon whereby a stressful but sublethal stimulus sets in motion a cascade of molecular and biochemical events that renders cells, tissues, or the whole organism tolerant to a future more lethal stimulus. Later, other groups attempted to apply the preconditioned responses to ischemia in neurologic injury models. Kitagawa et al. [99] showed that preconditioning with brief ischemia protected against a later injurious episode of bilateral common carotid artery occlusion in gerbils that otherwise caused damage to pyramidal neurons in the CA1 region of the hippocampus, a region of brain selectively vulnerable to ischemia. Subsequently, many groups have demonstrated that diverse preconditioning strategies can induce tolerance to global or focal brain ischemia.
Proinflammatory cytokines such as IL-1, TNF, and DAMPs activate intracellular signaling pathways that mediate stress responses and could, therefore, function in ligand–receptor interactions that induce a tolerant state. IL-1 has been observed to induce tolerance to global brain ischemia with protection of hippocampal CA1 neurons in the gerbil [100]. Bacterial lipopolysaccharide (LPS) is a potent stimulus for release of proinflammatory cytokines. Its administration in animal models has been reported to protect against ischemia in both the heart [101–103] and the brain [104]. Nitric oxide, heat shock protein 70, and superoxide dismutase have been implicated in LPS-induced tolerance to brain ischemia [105, 106]. Furthermore, LPS preconditioning has been shown to diverge the Toll-like receptor (TLR) 4 signaling cascade to suppress nuclear factor kappa B and to favor the activation of IFN regulatory factor (IRF) 3 to increase anti-inflammatory/type I IFN gene expression [107].
TLR2 and TLR9 have also been implicated in preconditioning. Preconditioning with the TLR2 ligand, Pam3CysSerLys4 (Pam3CSK4), 24 h prior to transient MCA occlusion significantly decreased infarct size and mortality while improving behavioral outcome in mice [108]. Similarly, preconditioning with the TLR9 ligand, unmethylated CpG oligodeoxynucleotides, was neuroprotective in transient MCA occlusion [109]. Like TLR4, TLR9 preconditioning increased TNF levels and TNF was required for neuroprotection [109].
Tolerance to severe ischemia induced by preconditioning fundamentally involves reprogramming of the genetic response to injury [110]. During exposure to severe ischemia, both a high percentage of unique transcripts and widespread, gene-specific repression of gene expression have been found in brain samples that had been preconditioned by sublethal ischemia compared with samples from nonpreconditioned brains. Also, the profiles of gene expression during severe ischemia appear to differ depending on the nature of the preconditioning stimulus, as shown for ischemic preconditioning versus LPS preconditioning. These preconditioning-specific phenotypes appear to be adapted to counter the specific cytotoxicity mechanisms of the preconditioning stimulus. This suggests that multiple nonoverlapping pathways may subserve tolerance to brain ischemia [110].
In an elegant study, Stevens et al. [111] compared the brain transcriptome in 3 preconditioning paradigms in mice: systemic administration of TLR4 or TLR9 ligands, and ischemic preconditioning. This study identified the type I IFN-regulated genes as a common denominator of all 3 preconditioning stimuli. Importantly, TLR preconditioning was abolished and ischemic preconditioning was reduced in mice with targeted deletion of IRF3 or IRF7, the 2 major transcription factors involved in the type I IFN response. These studies are important because they identify type I IFNs as a common downstream effector pathway involved in different preconditioning paradigms.
In addition, preconditioning protects cerebral blood vessels from the vasoparalysis induced by cerebral ischemia. Administration of LPS 24 h before MCA occlusion reduced ischemic brain injury and prevented the dysfunction in cerebrovascular regulation induced by MCA occlusion, as demonstrated by normalization of the increase in cerebral blood flow produced by neural activity, hypercapnia, or the endothelium-dependent vasodilator acetylcholine [112]. These beneficial effects of LPS were not observed in mice lacking inducible nitric oxide synthase or the NOX2 subunit of the superoxide-producing enzyme reduced nicotinamide adenine dinucleotide phosphate oxidase [112]. LPS increased ROS and the peroxynitrite marker 3-nitrotyrosine in cerebral blood vessels of wild-type mice but not in NOX2 nulls, and the peroxynitrite decomposition catalyst 5,10,15, 20-tetrakis(4-sulfonatophenyl)porphyrinato iron (III) counteracted the beneficial effects of LPS, which suggests that the vasoprotective effects of LPS are mediated by peroxynitrite produced by the reaction of inducible nitric oxide synthase-derived nitric oxide with NOX2-derived superoxide. As discussed by Kunz et al. [112], these findings are of interest because they indicate that peroxynitrite, in addition to its well-known deleterious vascular actions, can also protect cerebral blood vessels from the vascular dysregulation associated with cerebral ischemia.
Cell-based therapies are increasingly explored in preclinical and clinical settings for a variety of acute CNS injuries, including stroke [113–115]. While stem-cell therapies have been at the forefront of therapeutic efforts, there is increasing evidence that bone marrow mononuclear cells (BMMNCs) can confer protection in traumatic brain injury and stroke [116]. Because BMMNCs are heterogeneous and are composed of mature leukocytes, progenitors of the erythroid, myeloid, and lymphoid lineages, as well as hematopoietic and mesenchymal stem cells, it is difficult to attribute the neuroprotective effects to a defined cell population. A recent study found that BMMNCs depleted of myeloid cells lost their protective capacity in a model of transient MCA occlusion in mice, while lymphocyte or erythroid depletion had no effect [117]. The mechanisms by which BMMNCs confer neuroprotection remain elusive. Given that there is only minor engraftment of bone marrow-derived or cord blood cells into the ischemic tissue after intravenous delivery [118, 119], systemic immunomodulatory mechanisms should also be considered. Delivery of BMMNCs 24 h after transient MCA occlusion increased serum IL-10 and IFN-γ levels, while IL-1b, IL-6, and TNF were reduced [117, 120]. Vendrame et al. [118] reported that intravenously delivered umbilical cord cells in a stroke model in rats migrate to the spleen, restore splenic size, and prevent splenic CD8+ lymphocyte loss.
Conclusions
A growing body of evidence suggests that postischemic inflammation plays an important role in various stages of cerebral ischemic injury. The use of anti-inflammatory strategies in ischemic stroke therapy is attractive because they have a wider therapeutic window than the now-predominant approaches based on reperfusion. Studies in animal stroke models indicate that interventions aimed at attenuating the infiltration of certain leukocytes may have a beneficial effect in ameliorating the progression of ischemic brain damage. However, clinical trials that utilize antileukocyte agents have failed to show benefits [121]. Given that some lymphocytes, such as Treg or Th2 cells, might have beneficial effects in ischemic stroke, indiscriminate targeting of all leukocytes might not prove effective. In addition it becomes clear that inflammatory leukocytes, although detrimental during the acute phase of stroke, may transdifferentiate in situ to give rise to ant-inflammatory cells that by removing cell debris and secreting trophic factors might contribute to tissue repair processes during the chronic phase of cerebral ischemia. Recent work has also elucidated the bidirectional interaction of the peripheral immune system and the ischemic brain by identifying brain generated signals that lead to the peripheral immune response and the compensatory immunosuppression. In addition to the more traditional immune cell reservoirs, that is, bone marrow, spleen, and lymph nodes, the immune system of the intestinal tract has also been shown to participate in ischemic brain injury. Because the gut immune system can be skewed to a pro- or anti-inflammatory phenotype by altering the composition of intestinal microbiota, potential future strategies for postischemic immunomodulation might also involve targeting the gut bacteria.
References
Feigin VL, Norrving B, George MG, Foltz JL. Prevention of stroke: a strategic global imperative. Nat Rev Neurol 2016;12:501-512.
Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron 2010;67:181-198.
Fisher M, Saver JL. Future directions of acute ischaemic stroke therapy. Lancet Neurol 2015;14:758-767.
Jackman K, Iadecola C. Neurovascular regulation in the ischemic brain. Antioxid Redox Signal 2015;22:149-160.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med 2011;17:796-808.
De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke 2016;47:1165-1172.
Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory disequilibrium in stroke. Circ Res 2016;119:142-158.
Delvaeye M, Conway EM. Coagulation and innate immune responses: can we view them separately? Blood 2009;114:2367-2374.
Rezaie AR. Protease-activated receptor signalling by coagulation proteases in endothelial cells. Thromb Haemost 2014;112:876-882.
Amara U, Rittirsch D, Flierl M, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol 2008;632:71-79.
Széplaki G, Szegedi R, Hirschberg K, et al. Strong complement activation after acute ischemic stroke is associated with unfavorable outcomes. Atherosclerosis 2009;204:315-320.
Alawieh A, Elvington A, Tomlinson S. Complement in the homeostatic and ischemic brain. Front Immunol 2015;6:417.
Schäfer MK, Schwaeble WJ, Post C, et al. Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J Immunol 2000;164:5446-5452.
Mocco J, Mack WJ, Ducruet AF, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res 2006;99:209-217.
Cervera A, Planas AM, Justicia C, et al. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLOS ONE 2010;5:e8433.
Osthoff M, Katan M, Fluri F, et al. Mannose-binding lectin deficiency is associated with smaller infarction size and favorable outcome in ischemic stroke patients. PLOS ONE 2011;6:e21338.
Garcia-Bonilla L, Iadecola C. Peroxiredoxin sets the brain on fire after stroke. Nat Med 2012;18:858-859.
Benakis C, Garcia-Bonilla L, Iadecola C, Anrather J. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci 2014;8:461.
Singh V, Roth S, Veltkamp R, Liesz A. HMGB1 as a key mediator of immune mechanisms in ischemic stroke. Antioxid Redox Signal 2016;24:635-651.
Szalay G, Martinecz B, Lénárt N, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016;7:11499.
Huang J-M, Hu J, Chen N, Hu M-L. Relationship between plasma high-mobility group box-1 levels and clinical outcomes of ischemic stroke. J Crit Care 2013;28:792-797.
Liesz A, Mracsko E, Zhou W, et al. DAMP Signaling is a key pathway inducing immune modulation after brain injury. J Neurosci 2015;35:583-598.
Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab 2005;26:654-665.
Chapman KZ, Dale VQ, Denes A, et al. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J Cereb Blood Flow Metab 2009;29:1764-1768.
Ferrarese C, Mascarucci P, Zoia C, et al. Increased cytokine release from peripheral blood cells after acute stroke. J Cereb Blood Flow Metab 1999;19:1004-1009.
Basic Kes V, Simundic A-M, Nikolac N, Topic E, Demarin V. Pro-inflammatory and anti-inflammatory cytokines in acute ischemic stroke and their relation to early neurological deficit and stroke outcome. Clin Biochem 2008;41:1330-1334.
Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci 2005;6:775-786.
Prass K, Meisel C, Höflich C, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med 2003;198:725-736.
Langhorne P, Stott DJ, Robertson L, et al. Medical complications after stroke: a multicenter study. Stroke 2000;31:1223-1229.
Czlonkowska A, Cyrta B, Korlak J. Immunological observations on patients with acute cerebral vascular disease. J Neurol Sci 1979;43:455-464.
Offner H, Subramanian S, Parker SM, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol 2006;176:6523-6531.
Offner H, Vandenbark AA, Hurn PD. Effect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppression. Neuroscience 2009;158:1098-1111.
Seifert HA, Hall AA, Chapman CB, Collier LA, Willing AE, Pennypacker KR. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J Neuroimmune Pharmacol 2012;7:1017-1024.
Sahota P, Vahidy F, Nguyen C, et al. Changes in spleen size in patients with acute ischemic stroke: a pilot observational study. Int J Stroke 2013;8:60-67.
Courties G, Herisson F, Sager HB, et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res 2015;116:407-417.
Becker KJ. Sensitization and tolerization to brain antigens in stroke. Neuroscience 2009;158:1090-1097.
Urra X, Miró F, Chamorro A, Planas AM. Antigen-specific immune reactions to ischemic stroke. Front Cell Neurosci 2014;8:278.
Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015;523:337-341.
Ortega SB, Noorbhai I, Poinsatte K, et al. Stroke Induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov Med 2015;19:381-392.
Planas AM, Gómez-Choco M, Urra X, Gorina R, Caballero M, Chamorro A. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J Immunol 2012;188:2156-2163.
Korhonen P, Kanninen KM, Lehtonen S, et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav Immun 2015;49:322-336.
Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury And worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 2011;42:2026-2032.
Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007;27:2596-2605.
Schilling M, Strecker J-K, Ringelstein EB, Kiefer R, Schäbitz W-R. Turn-over of meningeal and perivascular macrophages in the brain of MCP-1-, CCR-2- or double knockout mice. Exp Neurol 2009;219:583-585.
Schilling M, Strecker J-K, Ringelstein EB, Schäbitz W-R, Kiefer R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain Res 2009;1289:79-84.
Denes A, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab 2008;28:1707-1721.
Kaito M, Araya S-I, Gondo Y, et al. Relevance of distinct monocyte subsets to clinical course of ischemic stroke patients. PLOS ONE 2013;8:e69409.
Urra X, Villamor N, Amaro S, et al. Monocyte subtypes predict clinical course and prognosis in human stroke. J Cereb Blood Flow Metab 2009;29:994-1002.
Gliem M, Mausberg AK, Lee J-I, et al. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol 2012;71:743-752.
Wattananit S, Tornero D, Graubardt N, et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 2016;36:4182-4195.
Hu X, Li P, Guo Y, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012;43:3063-3070.
Fumagalli S, Perego C, Ortolano F, De Simoni M-G. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia 2013;61:827-842.
Schilling M, Besselmann M, Müller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 2005;196:290-297.
Neher JJ, Emmrich JV, Fricker M, Mander PK, Théry C, Brown GC. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A 2013;110:E4098-E4107.
Sieber MW, Jaenisch N, Brehm M, et al. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLOS ONE 2013;8:e52982.
Felger JC, Abe T, Kaunzner UW, et al. Brain dendritic cells in ischemic stroke: time course, activation state, and origin. Brain Behav Immun 2010;24:724-737.
Gottfried-Blackmore A, Kaunzner UW, Idoyaga J, Felger JC, McEwen BS, Bulloch K. Acute in vivo exposure to interferon-gamma enables resident brain dendritic cells to become effective antigen presenting cells. Proc Natl Acad Sci U S A 2009;106:20918-20923.
Liesz A, Karcher S, Veltkamp R. Spectratype analysis of clonal T cell expansion in murine experimental stroke. J Neuroimmunol 2013;257:46-52.
Enzmann G, Mysiorek C, Gorina R, et al. The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol 2013;125:395-412.
del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke 1991;22:1276-1283.
Dawson DA, Ruetzler CA, Carlos TM, Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and microcirculatory perfusion in acute stroke in the SHR. Keio J Med 1996;45:248-252.
Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008;39:1121-1126.
Ludewig P, Sedlacik J, Gelderblom M, et al. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits MMP-9-mediated blood-brain-barrier breakdown in a mouse model for ischemic stroke. Circ Res 2013;113:1013-1022.
Forster C, Clark HB, Ross ME, Iadecola C. Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol 1999;97:215-220.
Garcia-Bonilla L, Moore JM, Racchumi G, et al. Inducible nitric oxide synthase in neutrophils and endothelium contributes to ischemic brain injury in mice. J Immunol 2014;193:2531-2537.
Garcia-Bonilla L, Racchumi G, Murphy M, Anrather J, Iadecola C. Endothelial CD36 contributes to postischemic brain injury by promoting neutrophil activation via CSF3. J Neurosci 2015;35:14783-14793.
Emerich DF, Dean RL, Bartus RT. The role of leukocytes following cerebral ischemia: pathogenic variable or bystander reaction to emerging infarct? Exp Neurol 2002;173:168-181.
Cuartero MI, Ballesteros I, Moraga A, et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARγ agonist rosiglitazone. Stroke 2013;44:3498-3508.
Mattila OS, Strbian D, Saksi J, et al. Cerebral mast cells mediate blood-brain barrier disruption in acute experimental ischemic stroke through perivascular gelatinase activation. Stroke 2011;42:3600-3605.
Strbian D, Kovanen PT, Karjalainen-Lindsberg M-L, Tatlisumak T, Lindsberg PJ. An emerging role of mast cells in cerebral ischemia and hemorrhage. Ann Med 2009;41:438-450.
Hurn PD, Subramanian S, Parker SM, et al. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab 2007;27:1798-1805.
Kleinschnitz C, Schwab N, Kraft P, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010;115:3835-3842.
Gelderblom M, Weymar A, Bernreuther C, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012;120:3793-3802.
Shichita T, Sugiyama Y, Ooboshi H, et al. Pivotal role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain injury. Nat Med 2009;15:946-950.
Benakis C, Brea D, Caballero S, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med 2016;22:516-523.
Gan Y, Liu Q, Wu W, et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci U S A 2014;111:2704-2709.
Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006;113:2105-2112.
Liesz A, Zhou W, Na S-Y, et al. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci 2013;33:17350-17362.
Liesz A, Suri-Payer E, Veltkamp C, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 2009;15:192-199.
Planas AM, Chamorro A. Regulatory T cells protect the brain after stroke. Nat Med 2009;15:138-139.
Li P, Gan Y, Sun B-L, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol 2013;74:458-471.
Kleinschnitz C, Kraft P, Dreykluft A, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood 2013;121:679-691.
Gill D, Veltkamp R. Dynamics of T cell responses after stroke. Curr Opin Pharmacol 2015;26:26-32.
Doyle KP, Quach LN, Solé M, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci 2015;35:2133-2145.
Roström B, Link B. Oligoclonal immunoglobulins in cerebrospinal fluid in acute cerebrovascular disease. Neurology 1981;31:590-596.
Prüss H, Iggena D, Baldinger T, et al. Evidence of intrathecal immunoglobulin synthesis in stroke: a cohort study. Arch Neurol 2012;69:714-717.
Abete P, Testa G, Cacciatore F, et al. Ischemic preconditioning in the younger and aged heart. Aging Dis 2011;2:138-148.
Offner H, Hurn PD. A novel hypothesis: regulatory b lymphocytes shape outcome from experimental stroke. Transl Stroke Res 2012;3:324-330.
Ren X, Akiyoshi K, Dziennis S, et al. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci 2011;31:8556-8563.
Nishio J, Honda K. Immunoregulation by the gut microbiota. Cell Mol Life Sci 2012;69:3635-3650.
Kuhn KA, Stappenbeck TS. Peripheral education of the immune system by the colonic microbiota. Semin Immunol 2013;25:364-369.
Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005;122:107-118.
Cebra JJ. Influences of microbiota on intestinal immune system development. Am J Clin Nutr 1999;69:1046S-1051S.
Bauer H, Horowitz RE, Levenson SM, Popper H. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am J Pathol 1963;42:471-483.
Winek K, Engel O, Koduah P, et al. Depletion of cultivatable gut microbiota by broad-spectrum antibiotic pretreatment worsens outcome after murine stroke. Stroke 2016;47:1354-1363.
Houlden A, Goldrick M, Brough D, et al. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun 2016;57:10-20.
Singh V, Roth S, Llovera G, et al. Microbiota Dysbiosis Controls the Neuroinflammatory Response after Stroke. J Neurosci 2016;36:7428-7440.
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986;74:1124-1136.
Kitagawa K, Matsumoto M, Tagaya M, et al. “Ischemic tolerance” phenomenon found in the brain. Brain Res 1990;528:21-24.
Ohtsuki T, Ruetzler CA, Tasaki K, Hallenbeck JM. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J Cereb Blood Flow Metab 1996;16:1137-1142.
Song W, Furman BL, Parratt JR. Delayed protection against ischaemia-induced ventricular arrhythmias and infarct size limitation by the prior administration of Escherichia coli endotoxin. Br J Pharmacol 1996;118:2157-2163.
Eising GP, Mao L, Schmid-Schönbein GW, Engler RL, Ross J. Effects of induced tolerance to bacterial lipopolysaccharide on myocardial infarct size in rats. Cardiovasc Res 1996;31:73-81.
Rowland RT, Meng X, Cleveland JC, Meldrum DR, Harken AH, Brown JM. LPS-induced delayed myocardial adaptation enhances acute preconditioning to optimize postischemic cardiac function. Am J Physiol 1997;272:H2708-H2715.
Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res 1997;748:267-270.
Puisieux F, Deplanque D, Pu Q, Souil E, Bastide M, Bordet R. Differential role of nitric oxide pathway and heat shock protein in preconditioning and lipopolysaccharide-induced brain ischemic tolerance. Eur J Pharmacol 2000;389:71-78.
Bordet R, Deplanque D, Maboudou P, et al. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J Cereb Blood Flow Metab 2000;20:1190-1196.
Vartanian KB, Stevens SL, Marsh BJ, Williams-Karnesky R, Lessov NS, Stenzel-Poore MP. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflamm 2011;8:140.
Hua F, Ma J, Ha T, et al. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol 2008;199:75-82.
Stevens SL, Ciesielski TMP, Marsh BJ, et al. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab 2008;28:1040-1047.
Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke 2007;38(2 Suppl.):680-685.
Stevens SL, Leung PY, Vartanian KB, et al. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci 2011;31:8456-8463.
Kunz A, Park L, Abe T, et al. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 2007;27:7083-7093.
George PM, Steinberg GK. Novel stroke therapeutics: unraveling stroke pathophysiology and its impact on clinical treatments. Neuron 2015;87:297-309.
Rosado-de-Castro PH, Pimentel-Coelho PM, Barbosa da Fonseca LM, de Freitas GR, Mendez-Otero R. The rise of cell therapy trials for stroke: review of published and registered studies. Stem Cells Dev 2013;22:2095-2111s.
Bang OY. Clinical trials of adult stem cell therapy in patients with ischemic stroke. J Clin Neurol 2016;12:14-20.
Vahidy FS, Rahbar MH, Zhu H, Rowan PJ, Bambhroliya AB, Savitz SI. Systematic review and meta-analysis of bone marrow-derived mononuclear cells in animal models of ischemic stroke. Stroke 2016;47:1632-1639.
Yang B, Parsha K, Schaar K, Xi X, Aronowski J, Savitz SI. Various cell populations within the mononuclear fraction of bone marrow contribute to the beneficial effects of autologous bone marrow cell therapy in a rodent stroke model. Transl Stroke Res 2016;7:322-330.
Vendrame M, Gemma C, Pennypacker KR, et al. Cord blood rescues stroke-induced changes in splenocyte phenotype and function. Exp Neurol 2006;199:191-200.
Borlongan CV. Central nervous system entry of peripherally injected umbilical cord blood cells is not required for neuroprotection in stroke. Stroke 2004;35:2385-2389.
Yang B, Migliati E, Parsha K, et al. Intra-arterial delivery is not superior to intravenous delivery of autologous bone marrow mononuclear cells in acute ischemic stroke. Stroke 2013;44:3463-3472.
Veltkamp R, Gill D. Clinical trials of immunomodulation in ischemic stroke. Neurotherapeutics 2016; doi:10.1007/s13311-016-0458-y.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Anrather, J., Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 13, 661–670 (2016). https://doi.org/10.1007/s13311-016-0483-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0483-x