
Abstract
Herpetic infections have plagued humanity for thousands of years, but only recently have advances in antiviral medications and supportive treatments equipped physicians to combat the most severe manifestations of disease. Prompt recognition and treatment can be life-saving in the care of patients with herpes simplex-1 virus encephalitis, the most commonly identified cause of sporadic encephalitis worldwide. Clinicians should be able to recognize the clinical signs and symptoms of the infection and familiarize themselves with a rational diagnostic approach and therapeutic modalities, as early recognition and treatment are key to improving outcomes. Clinicians should also be vigilant for the development of acute complications, including cerebral edema and status epilepticus, as well as chronic complications, including the development of autoimmune encephalitis associated with antibodies to the N-methyl-D-aspartate receptor and other neuronal cell surface and synaptic epitopes. Herein, we review the pathophysiology, differential diagnosis, and clinical and radiological features of herpes simplex virus-1 encephalitis in adults, including a discussion of the most common complications and their treatment. While great progress has been made in the treatment of this life-threatening infection, a majority of patients will not return to their previous neurologic baseline, indicating the need for further research efforts aimed at improving the long-term sequelae.
Similar content being viewed by others

Introduction
Encephalitis is inflammation of the brain parenchyma with neurologic dysfunction, and can result from infectious, postinfectious, and noninfectious causes [1]. Infection constitutes approximately 50 % of identifiable cases and is the most commonly identified etiologic category of encephalitis [2]. Herein, we review the clinical and radiological manifestations, diagnostic evaluation, and treatment of herpes simplex virus-1 (HSV-1) encephalitis (HSVE), the most common infectious cause of sporadic encephalitis.
Herpetic infections have been recognized since the time of ancient Greece. The word herpes translates as “creeping” or “crawling”, and is a reference to herpetic skin lesions. Goodpasture [3] and others demonstrated that material from herpetic lip and genital lesions produced encephalitis when introduced into the scarified cornea or skin of rabbits. In the 1920s, the Mathewson commission was among the earliest reports to suggest HSV caused encephalitis in humans [4]. The first pediatric case report of HSVE was published in 1941 [5]. The first adult case, a 25-year-old man who presented with headache, fever, aphasia, and left pupillary dilatation, was reported in 1944 [6]. On postmortem pathological examination, there were numerous petechiae and ecchymoses with perivascular lymphocytic cuffing in the left temporal lobe, midbrain, and pons. Intranuclear inclusions were identified and virus was isolated from the patient’s brain. Significant progress in the pathobiology, diagnosis, and treatment of HSVE has been made since these early reports.
Pathophysiology
HSV-1 is 1 of 8 human herpes viruses (HHV), including HSV-2, varicella zoster virus (VZV; HHV-3), Epstein–Barr virus (HHV-4), cytomegalovirus (HHV-5), HHV-6, HHV-7, and HHV-8. The herpesviruses are large, double-stranded DNA viruses that are well-adapted to human infection as they establish lifelong infection, rarely cause death of the host, and are readily spread between individuals.
HSV initially gains access to host tissues through mucous membranes or damaged skin. After primary infection of the mucosal or skin epithelium, the virus infects sensory neurons via interactions with cell-surface glycosaminoglycans such as heparan sulfate [7], and cell adhesion molecules such as nectin-1 [8, 9], and travels to the neuronal cell body in the dorsal root ganglion via fast retrograde axonal transport [10, 11].
The mechanisms by which HSV gains access to the central nervous system (CNS) in humans are unclear, and this remains an area of debate. The most likely routes include retrograde transport through the olfactory or trigeminal nerves [9, 12, 13], or via hematogenous dissemination. The viral tropism for the orbitofrontal and mesiotemporal lobes argues against hematogenous dissemination in most cases. Experimental evidence in animals supports transmission to the CNS via either or both the trigeminal and olfactory routes, and suggests that virions can spread to the contralateral temporal lobe via the anterior commissure [13].
Unlike other cranial nerves with sensory functions, the olfactory nerve pathways do not route through the thalamus but connect directly to the frontal and mesiotemporal lobes (including the limbic system). There is some evidence to support olfactory spread to the CNS in humans, but definitive data are lacking [12, 14–16]. The trigeminal nerve innervates the meninges, and spread to the orbitofrontal and mesiotemporal lobes could also occur through this route [17]. However, as the sensory nuclei of the trigeminal nerve are located in the brainstem, one would expect the relatively rare occurrence of brainstem encephalitis associated with HSVE to be more common if this were the primary route of entry to the CNS in most cases [18–20].
Whether HSVE is a reactivation of latent virus or caused by primary infection is also an area of contention; both may occur. Proposed pathogenic mechanisms include reactivation of latent HSV in the trigeminal ganglia with subsequent spread of infection to the temporal and frontal lobes, primary CNS infection, or perhaps reactivation of latent virus within the brain parenchyma itself [17, 21–23]. In at least half of HSVE cases, the viral strain responsible for encephalitis is different from the strain that causes herpetic skin lesions in the same patient, an observation that suggests the possibility of primary CNS infection [24].
Infection with HSV triggers a robust response from the innate immune system until adaptive immunity is able to assist in clearing active infection. Early in the course of the immune response to HSV, pattern recognition receptors, called Toll-like receptors (TLRs), located on cells of the innate immune system, recognize and bind to conserved viral motifs known as pathogen associated molecular patterns [25]. This triggers dimerization of the TLRs, which subsequently activates signaling pathways that initiate the production of proinflammatory cytokines such as interferons (IFNs), tumor necrosis factor, and various interleukins [26]. IFNs contribute to host resistance to viral proliferation through activation of the Jak-Stat signaling pathway [27], and by triggering production of both RNAse enzymes that destroy cellular RNA (both host and viral) and double-stranded RNA-dependent protein kinase, which halts cellular translation [28]. Deficiencies in the immune response to HSV (e.g., defects in the TLR-3 pathway, including TLR3 itself, UNC93B1, TIR-domain-containing adapter-inducing IFN-β, tumor necrosis factor receptor-associated factor-3, TANK-binding kinase 1, or IFN regulatory factor-3) leave the host susceptible to HSVE [29–31].
The inflammatory cascade recruits innate immune cells and primes adaptive immunity, which can lead to necrosis and apoptosis of infected cells. While the host immune response is critical to eventual viral control, the inflammatory response, particularly recruitment of activated leukocytes, may contribute to tissue destruction and consequent neurologic sequelae [32, 33].
After primary infection, the virus establishes a latent state for the life of the host and remains quiescent unless reactivated [34]. In order to establish and maintain latency, a number of complex processes must be balanced. These include silencing of viral lytic-phase genes, abrogation of host cellular defense mechanisms (e.g., apoptosis), and evasion of host immunity, including both innate and acquired immune responses (e.g., suppression of major histocompatibility complex expression) [35, 36]. HSV-specific CD8+ T cells take up residence in the trigeminal ganglia and contribute to maintaining the virus in the latent state [37]. During reactivation, the expression of viral genes occurs in a temporally organized fashion, as reviewed recently [38]. Once reactivated, the virus can infect neighboring neurons and travel to tissues innervated by the infected dorsal root ganglia to cause recurrent disease and shed infectious viral particles that can be transmitted to others.
Epidemiology
HSV-1 infection is common, with seropositivity among older adults estimated to be 60–90 % worldwide [39]. A survey from 2005 to 2010 including Americans between 14 and 49 years of age in the USA estimated HSV-1 seropositivity at ~54 % and HSV-2 seropositivity at ~16 % [40]. While HSV-2 is also capable of causing encephalitis (particularly in the immunocompromised host), HSV-1 is responsible for ~90 % of HSV encephalitis in adults and children, and is the focus of this review [41]. Despite only rarely manifesting as encephalitis in infected individuals, HSV-1 is consistently the single most common cause of sporadic encephalitis worldwide [42–52]. The incidence of HSVE worldwide is estimated to be between 2 and 4 cases/1,000,000 [44], and the incidence in the USA is similar [53]. There is a bimodal distribution with peak incidence in the very young (up to 3 years of age), and again in adults aged > 50 years, but the majority of cases occur in those over 50, with both sexes equally affected [44, 54–56].
Clinical Manifestations
Key to early recognition and treatment of HSVE is familiarity with the syndrome of encephalitis, which includes altered mental status (typically for ≥ 24 h), accompanied by evidence of brain parenchymal inflammation. Findings supportive of brain inflammation may include fever, new seizures, focal neurologic signs, cerebrospinal fluid (CSF) pleocytosis (≥5 nucleated cells/ml), and radiological and/or neurophysiologic abnormalities [e.g., contrast-enhancing lesions on magnetic resonance imaging (MRI) or abnormal findings on electroencephalography (EEG), respectively] [1]. Encephalitis must be distinguished from encephalopathy, a broader term that refers to a clinical state of disorientation, confusion, behavioral, and other cognitive changes that can occur in the setting of encephalitis, as well as numerous other noninflammatory conditions.
Many patients present with prodromal symptoms, suggesting upper respiratory tract or other systemic infection. Signs and symptoms of encephalitis then progress over the course of several days in most cases of HSVE [57, 58]. The most common manifestations include encephalopathy, fever, seizures, headaches, and focal neurological deficits [57–62]. Although clinical features of HSVE have been well described in multiple large epidemiological studies, the clinical manifestations lack specificity. In a series of 106 cases of HSVE, the primary reasons for hospital presentation were seizures (32 %), abnormal behavior (23 %), loss of consciousness (13 %), and confusion or disorientation (13 %) [60].
Immunocompromised Individuals
Immunocompromised patients present a greater diagnostic challenge. In the largest series to date, Tan et al. [63] retrospectively reviewed and compared the clinical manifestations and course of immunocompromised and immunocompetent patients with HSVE. In that study, immunocompromised patients were less likely to present with prodromal symptoms or with focal neurologic deficits. They had more extensive brain involvement that was more often distributed outside the temporal lobes and it was not uncommon to observe a lack of pleocytosis in the CSF. Morbidity and mortality were significantly higher in the immunocompromised group, with 35.7 % mortality compared with 6.7 % mortality in the immunocompetent. Autopsy in 3 immunocompromised patients who died of HSVE revealed an atypical, noninflammatory, “pseudoischemic” histologic pattern [64].
Evaluation and Differential Diagnosis
In the setting of suspected encephalitis, the value of a thorough history and physical examination cannot be overstated, and a thoughtful approach is critical to narrowing the differential. Key elements of the history are intended to identify alternative causes of encephalitis and include immune-suppressing medications or illness, travel history (both recent and remote), and mosquito/tick exposure. Weight loss and infectious symptoms, including low-grade fever, rash, and so on, and neurologic or psychiatric abnormalities such as aphasia, behavioral changes and seizure-like activity should also be reviewed. Full neurologic and general medical examinations are critical and may uncover clues to the diagnosis. Patterns of neurologic dysfunction may help to suggest an etiology, for example cranial neuropathies and autonomic instability may suggest a brainstem encephalitis, which can help to narrow the differential diagnosis [65]. Tremors, movement disorders, or other signs referable to the basal ganglia may also assist in guiding the differential [65]. Differentiating encephalitis from its mimics can be especially challenging in the elderly and the immunocompromised. Focused laboratory testing and prompt neuroimaging assist greatly in the diagnostic approach.
Laboratory Studies
Serum laboratory studies that should be obtained on all adults with encephalitis include complete blood count with differential, electrolytes, measures of renal and liver function, blood cultures, HIV testing (consider RNA), and treponemal testing. In children with encephalitis, Mycoplasma pneumoniae IgM and IgG, as well as Epstein–Barr virus serologies (VCA IgG and IgM and EBNA IgG), should be obtained. Serum should also be reserved from the presentation, with convalescent serum collected 10–14 days later for paired antibody testing if needed (such as in idiopathic encephalitis). HSV serologies are generally not clinically helpful in the acute setting [66]. In patients at risk for tuberculosis, such as the immunocompromised and homeless individuals, skin or blood testing for Mycobacterium tuberculosis should be considered.
Unless contraindicated (see acute complications; edema), lumbar puncture should be obtained in all patients with encephalitis, but should not delay the administration of empiric antimicrobials. Important studies to obtain in adults with encephalitis include opening pressure, cell count and differential, protein, glucose, Gram stain, oligoclonal bands, IgG index, bacterial cultures, HSV-1/HSV-2 polymerase chain reaction (PCR), VZV PCR (and IgG and IgM if available), enterovirus PCR, cryptococcal antigen or India ink staining, and Venereal Disease Research Laboratory test. The opening pressure in HSVE is generally normal or slightly elevated. There is considerable variation in the CSF profile of HSVE, but a typical profile includes a moderate lymphocytic pleocytosis (10–200/mm3), may demonstrate elevated erythrocytes (normal–minimally elevated counts are common), moderately elevated protein (50–100 mg/dl), and normal glucose (although hypoglycorrhachia may be present in a minority of patients) [60]. PCR for HSV-1 and HSV-2, which has supplanted viral cultures and other studies as the test of choice, should be obtained from the CSF and has high sensitivity (96 %) and specificity (99 %) [67, 68]. False-negative PCR can occur early in the illness [98–100], and if the clinical suspicion is high, aciclovir should be continued empirically and repeat CSF HSV PCR obtained within 3–7 days [43].
Neuroimaging
Computed tomographic (CT) imaging is generally inadequate for the evaluation of encephalitis, but, in practice, is often obtained as the initial neuroimaging study in the encephalopathic patient and may suggest an alternate etiology. CT imaging in encephalitis is beneficial for rapid evaluation of patients in whom there is clinical concern for edema and/or shift of brain compartments that might require intervention or contraindicate lumbar puncture. Abnormal findings have been observed in 25–80 % of patients with HSVE imaged soon after admission [62, 69]. CT findings suggestive of HSVE include hypodense lesions (typically in the temporal lobe), edema, or contrast enhancement [70–72]. However, CT is unable to differentiate between HSVE and many of its mimics, and lacks sensitivity, particularly early in the course of the illness. For diagnostic purposes, MRI is superior to CT. For example, in a recent study [60], CT scan was abnormal in roughly half of all cases, while MRI was abnormal in nearly all patients with HSVE.
MRI with and without contrast is the neuroimaging study of choice in the evaluation of encephalitis and is abnormal in the vast majority of cases of HSVE [73]. MRI is the most sensitive and specific imaging method for HSVE, particularly early in the course of the illness [74]. Typical findings on MRI include asymmetric hyperintense lesions on T2-weighted sequences corresponding to areas of edema in the mesiotemporal and orbitofrontal lobes and the insular cortex [75].
Accumulating evidence suggests that diffusion restriction on diffusion-weighted imaging (DWI) is frequently seen early in the course of HSVE and may be among the earliest neuroradiologic manifestations [76]. McCabe et al. [77] reported an adult with HSVE in whom HSV PCR was negative, but early diffusion restriction was observed in the anterior temporal lobes and the insular cortex. More reports demonstrating improved sensitivity of DWI over fluid-attenuated inversion recovery (FLAIR) sequences were soon to follow in adults [78], children [79], and neonates [80, 81]. One report demonstrated correlation between DWI lesions and clinical response to treatment [82]. In the largest retrospective study to date comparing DWI with FLAIR, Renard et al. [83] demonstrated that early in the course (<2 weeks from symptom onset) DWI demonstrated as many or more lesions, and these were easier to visualize compared with FLAIR. FLAIR signal abnormalities appeared more prominent later in the course. The authors also noted early signal changes in the thalamus that were detected on FLAIR but not DWI—a finding that appeared to be related to HSVE and not associated seizures or other factors. Overall, DWI changes in the temporal or frontal lobes in the appropriate clinical setting should be considered a clue to the diagnosis of HSVE.
While traditional teaching has emphasized bilateral temporal involvement as characteristic of HSVE, this has not held true in contemporary studies. On the contrary, a recent study of cases of encephalitis with temporal lobe abnormalities found that bilateral temporal lobe involvement was associated with lower odds of HSVE compared with all other etiologies and when directly compared with autoimmune etiologies [84]. In that study of immune competent adults, patients with HSVE, as compared with other infectious or autoimmune etiologies of their temporal lobe encephalitis, were more likely to be older and white, and to present acutely and with fever, seizures, and upper respiratory symptoms. In addition to bilateral temporal lobe involvement, lesions outside the temporal lobe or limbic region suggested an alternate diagnosis.
EEG
In the acute setting, a number of electrographic findings have been associated with HSVE, including periodic discharges, focal or generalized slowing, and electrographic seizures, including status epilepticus (SE) [85, 86]. Seizures and epilepsy in the setting of HSVE have recently been reviewed [87]. Periodic discharges in HSVE have been observed generally between days 2 and 15 and may manifest before structural lesions can be observed on CT [88, 89]. While EEG is recommended as part of the diagnostic evaluation of patients with encephalitis, there are few studies characterizing the contribution of EEG to diagnosis and prognosis in these patients, particularly in the era of MRI. Sutter et al. [90] recently reviewed 103 patients with encephalitis who presented between 1997 and 2011, 12 of whom had HSVE [90]. Patients with HSVE were significantly more likely to have periodic discharges and focal slowing in the frontotemporal and occipital areas compared with patients with encephalitis of other etiologies, consistent with previous studies [91, 92].
Differential Diagnosis
In 1989, Whitley et al. [93] reviewed 432 cases of encephalitis that underwent temporal lobe biopsy for presumed HSVE. Among these, 195 patients (45 %) had HSVE, 95 patients (22 %) had other identifiable etiologies, and 143 patients (33 %) remained idiopathic, despite biopsy. Among the most common treatable mimics were other infections (viral, bacterial, mycobacterial, and fungal), malignancy, vascular disease (more often hemorrhagic than thrombotic), and a few cases of toxic or metabolic disease. No diagnostic studies, alone or in combination, were felt to be sufficiently characteristic to be diagnostically useful. Since that study, the advent of MRI and establishment of HSV PCR (see above) have significantly improved the clinician’s ability to diagnose HSVE. However, even with contemporary diagnostic modalities, the identification of HSVE mimics remains challenging. Numerous infectious agents and autoimmune syndromes may present similarly to HSVE. In addition, a number of other conditions can mimic HSVE (Table 1).
Chow et al. [84] recently reviewed the clinical and neuroimaging features of 251 cases of temporal lobe encephalitis from the California Encephalitis Project. Among all cases of temporal lobe encephalitis, 43 % were infectious and 16 % were noninfectious etiologies. Of the infectious causes, HSVE was the most commonly identified agent, followed by tuberculosis and VZV. In the absence of zoster, HSV and VZV can be clinically indistinguishable [94]. More than half of the noninfectious etiologies were immune-mediated, including anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis, antivoltage-gated potassium channel complex encephalitis (more precisely anti-leucine-rich glioma-inactivated protein 1 [LGI1] and anti-contactin- associated protein-like 2 [Caspr2] antibodies), and CNS vasculitis. Despite extensive evaluation, 41 % remained idiopathic.
Diagnostic Pitfalls
There are 3 things to consider. 1) Failure to recognize encephalitis. This can lead to insufficient testing (i.e., not obtaining MRI and CSF studies which can lend support to the diagnosis). 2) Absence of CSF pleocytosis. As noted above, multiple studies have demonstrated that immunocompromised patients are less likely to have CSF pleocytosis [63, 95–97]. 3) False-negative PCR studies. HSV-1 PCR may yield a false-negative, particularly early in the course of the HSVE and among children [98–100]. When suspicion is high, patients should be treated empirically, despite a negative PCR, and HSV PCR from the CSF should be repeated within 3–7 days [43].
Management
Initial Management
The first priority on presentation is to recognize and treat any emergent issues (Fig. 1). This includes rapid evaluation of hemodynamic and respiratory sufficiency, which is particularly important in the setting of decreased level of consciousness. Rapid evaluation for other potentially reversible causes of encephalopathy such as hypoglycemia, hypercarbia, electrolyte abnormalities, and so on, can readily be performed in the emergency setting, and abnormalities should be treated promptly. After initial stabilization, the patient should be appropriately triaged and may require admission to the intensive care unit (ICU) [101]. Decreased level of consciousness, severe comorbidities, and autonomic dysfunction are some of the indications for ICU admission. Whenever possible, a dedicated neurological ICU is recommended; barring this, admission to a medical ICU or rapid transportation to the closest neurological ICU should be considered. Close real-time coordination of care with a multidisciplinary medical team (i.e., critical care, neurology, and infectious disease) is suggested.

Management of patients with suspected herpes simplex virus-1 encephalitis (HSVE). Adapted from Venkatesan and Geocadin [101]. cEEG = continuous electroencephalography; CSF = cerebrospinal fluid; ICP = intracranial pressure; ICU = intensive care unit; PCR = polymerase chain reaction; SE = status epilepticus
Empirical Treatment of Encephalitis
While clinical, laboratory, radiographic, and neurophysiologic findings on presentation may suggest HSVE, no combination of features is sufficiently sensitive and empirical treatment should be initiated in all patients with encephalitis [43]. As noted below, early initiation of aciclovir is the most readily modifiable factor for improving outcomes. Intravenous aciclovir 10 mg/kg q8h for 14–21 days should, therefore, be initiated promptly and the diagnostic evaluation should never delay antimicrobial therapy in patients with encephalitis [43]. As bacterial meningoencephalitis often cannot be excluded on clinical grounds, and septic encephalopathy is a common mimic of HSVE [102], we recommend the addition of broad-spectrum antibiotics until bacterial infection can be excluded. Recent UK guidelines for the empiric management of encephalitis support this approach [65]. Following initiation of antimicrobial therapy, continued close clinical evaluation and frequent revisiting of the possibility of alternate diagnoses can help to avoid premature closure when a diagnosis has not yet been established.
Antiviral Medication
IV aciclovir is the first-line treatment for HSVE at a dose of 10 mg/kg q8h and should be continued for 14–21 days (Table 2). The benefit of aciclovir in HSVE was established by 2 landmark clinical trials conducted in the mid-1980s. Whitley et al. [103] randomized 208 patients with presumed HSVE to either aciclovir intravenously at a dose of 10 mg/kg q8h or vidarabine for 10 days. All patients underwent diagnostic brain biopsy, among whom 69 (33 %) had biopsy-proven HSVE. Treatment with aciclovir was associated with a significantly reduced rate of mortality compared with vidarabine (28 % vs 54 %; p = 0.008). This supported the results from a multicenter Swedish study published in 1984 that compared aciclovir with vidarabine in 127 patients with presumed HSVE (53 of whom had biopsy-confirmed HSVE) [104]. That study also found that aciclovir treatment reduced mortality compared with vidarabine (19 % vs 50 %; p = 0.04). Together, these trials established aciclovir as the standard of care in HSVE.
Aciclovir is a nucleoside analog with strong antiviral activity against HSV-1, HSV-2, and VZV, and is a relatively safe medication. After uptake into the cell, virally encoded thymidine kinase phosphorylates aciclovir into aciclovir monophosphate, which is subsequently phosphorylated into the active aciclovir triphosphate by cellular kinases. The initial phosphorylation of aciclovir does not occur to any appreciable extent in noninfected cells, providing a degree of selectivity for infected cells. An analog to deoxyguanosine triphosphate, aciclovir triphosphate competitively inhibits viral DNA polymerase and is incorporated into DNA, which leads to chain termination as a result of the absence of a 3' hydroxyl moiety. The affinity of aciclovir triphosphate is much higher for viral DNA polymerase than for the human homolog, which increases the therapeutic window [105, 106].
Oral aciclovir is approximately 15–30 % bioavailable and achieves widespread tissue and fluid penetration with CSF concentration roughly 50 % of that in serum. The plasma half-life is approximately 2–3 h in patients with normal renal function but is longer in those with renal insufficiency, for whom doses must be reduced. Patients with creatinine clearance (CrCl) of 25–50 ml/min/1.73 m2 should be given 10 mg/kg q12h; those with CrCl 10–25 ml/min/1.73 m2, 10 mg/kg q24h; and those with CrCl < 10 ml/min/1.73 m2 5 mg/kg q24h. Patients on thrice-weekly hemodialysis should be given 2.5–5.0 mg/kg q24h (given after dialysis on those days), while those on peritoneal dialysis should be treated with 10 mg/kg q24h [107]. Approximately 15 % (9–22 % in 1 study [108]) of drug is bound to serum proteins and therefore much of the drug can be removed by dialysis. No dosage adjustments are needed in patients with hepatic impairment.
Aciclovir is cleared by both glomerular filtration and tubular secretion and can precipitate in the renal tubules to cause obstructive nephropathy [109]. When this occurs, it typically develops after several days of therapy and may affect as many as 20 % of patients but is generally reversible [110]. Given this risk, we routinely monitor renal function, provide a slow infusion over 1–2 h, and pretreat with intravenous fluids to maintain urine output of approximately 75 ml/h. Neurotoxicity is rarely reported, mostly in patients with pre-existing renal insufficiency, and manifests as delirium, tremors, myoclonus, and possibly coma [111]. This can be difficult to diagnose in the setting of HSVE. Given the risks of toxicity, doses of aciclovir should be reduced as appropriate in patients with pre-existing renal insufficiency, particularly those on dialysis.
Aciclovir is considered pregnancy category B by the US Food and Drug Administration, indicating no clear risk in humans. At least 1 large observational study which included 1804 pregnancies with exposure to aciclovir, valaciclover, or famciclovir during the first trimester demonstrated no correlation between exposure and an increased risk of birth defects [112].
Although rare (i.e., < 1 % in the immunocompetent), viral resistance to aciclovir has emerged, particularly among patients with immunocompromise [113], and may be encountered in as many as 30 % of patients who have undergone bone marrow transplantation, who appear to be the highest-risk group. Treatment resistance should be considered in patients who are not responding as expected to standard treatment, or when there is evidence of clinical worsening, though it can be difficult to determine whether this represents treatment failure or the natural course of the illness. Three primary mechanisms of viral resistance to aciclovir have been described: absent or decreased levels of thymidine kinase, decreased phosphorylation of aciclovir by thymidine kinase, and decreased affinity of viral DNA polymerase for aciclovir triphosphate [114, 115].
Viral resistance to aciclovir can be associated with resistance to other antiviral tyrosine kinase-dependent nucleoside analogs such as ganciclovir, penciclovir, and its prodrug, famciclovir. In the case of aciclovir resistance, the preferred treatment is foscarnet [116, 117], a pyrophosphate analog and selective inhibitor of viral DNA polymerase that does not require phosphorylation for its antiviral activity [118–120]. After binding to viral DNA polymerase, foscarnet prevents the cleavage of the pyrophosphate moiety from deoxynucleotide triphosphates, thereby abridging chain elongation.
As foscarnet has poor oral bioavailability, it is given intravenously and approximately 20 % of administered drug is taken up by bone and cartilage. The drug undergoes minimal metabolism and is almost exclusively cleared by both glomerular filtration and tubular secretion [120]. The dosing in HSVE is 90 mg/kg i.v. q12h or 60 mg/kg i.v. q8h and should be reduced for patients with renal insufficiency.
Foscarnet is more toxic than aciclovir and should be given in consultation with experts in infectious disease. Renal toxicity resulting from direct tubular injury can be attenuated by giving intravenous fluids concomitantly [121]. Electrolyte abnormalities including hypocalcemia and hypomagnesemia are another common occurrence during treatment, and may contribute to reports of seizures associated with foscarnet treatment [120]. Nausea is also common during infusion. As with aciclovir, monitoring of electrolytes and renal function is important during foscarnet administration.
In the event of aciclovir shortage, ganciclovir or foscarnet can be given. Ganciclovir is an analog of the nucleoside guanosine that is activated by viral and cellular kinases to the triphosphate form, which then preferentially inhibits viral DNA polymerase, similar to aciclovir [122]. The drug is excreted unmodified in the urine and dose reduction is necessary in patients with renal insufficiency. The dosing for ganciclovir is 5 mg/kg q12h. Cidofovir should not be given for infections of the CNS, however, as it achieves inadequate penetration of the blood–brain barrier.
Duration of Treatment
The current guidelines recommend intravenous aciclovir for 14–21 days in cases of HSVE [43]. Though the initial studies provided aciclovir for 10 days, relapses beyond this were subsequently reported [123, 124], prompting most physicians to provide a longer duration of therapy (notably, many cases of apparent relapse may be immune mediated rather than infectious—see below). Some advocate repeat CSF PCR at 14–21 days, with longer treatment if PCR is positive [125, 126], though we generally reserve this for patients who have had a complicated course or have not responded to therapy as well as expected. A recently completed trial noted a lack of benefit from an additional 3 months of oral valaciclovir for the reduction of post-HSVE neuropsychiatric complications in 87 adults with PCR-proven HSVE [127].
According to the UK guidelines for the treatment of encephalitis, aciclovir can be safely discontinued in immunocompetent patients when an alternative diagnosis is established, or HSV PCR from the CSF has been negative on 2 occasions at least 24–48 h apart, or if all of the following conditions are met: negative CSF PCR obtained > 72 h from symptom onset, no alteration of consciousness, normal brain MRI, and CSF leukocytes are < 5 cells/ml [65].
Corticosteroids
Preclinical and animal studies have suggested a potential benefit associated with the use of corticosteroids in HSVE [128]; however, clinical evidence in humans is scant. While the host immune system paradoxically contributes to tissue injury, it is also important for suppressing viral spread and replication. As corticosteroids have both potent anti-inflammatory and immunomodulatory effects that may, theoretically, facilitate viral replication, it is not surprising that differing opinions exist regarding their use in HSVE [129, 130]. A nonrandomized retrospective study of 45 patients with HSVE suggested that the addition of corticosteroids to aciclovir may be associated with improved outcomes [131], thus prompting larger-scale clinical trials.
The German trial of aciclovir and corticosteroids in HSVE (GACHE) was a multicenter, multinational randomized, placebo-controlled clinical trial intended to compare aciclovir plus dexamethasone to aciclovir and placebo [132]. Patients with CSF HSV PCR positivity were to be randomized to the experimental or control group. Both groups would be given aciclovir 10 mg/kg q8h for 14 days. The experimental arm of the trial would receive 40 mg dexamethasone q24h for 4 days [133]. However, the trial was not completed as a result of limited recruitment.
The dexamethasone in herpes simplex virus encephalitis (DEX-ENCEPH) trial is a multinational, randomized controlled trial that is currently enrolling patients with HSVE with CSF PCR positivity. Patients will be randomized to receive dexamethasone 10 mg q6h for 4 days or no steroids, and the primary outcome will be a verbal memory score. The UK encephalitis guidelines have suggested against the routine use of corticosteroids in HSVE until results from controlled trials are available [65]. Our practice has been to reserve corticosteroids for patients in whom there is significant edema and mass effect.
Complications of HSVE
In addition to respiratory and circulatory insufficiency, important acute neurologic complications of encephalitis include seizures and elevated intracranial pressure associated with brain edema and herniation.
Case 1: A 33-Year-old Woman with Complications of HSVE
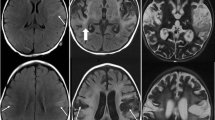
A 33-year-old woman presented with a generalized tonic-clonic seizure after several days of upper respiratory tract infection, headache, fever, confusion, and word-finding difficulty. Brain MRI revealed diffusion restriction and FLAIR hyperintensity with edema in the mesial temporal lobe and hypothalamus (Fig. 2). EEG demonstrated periodic discharges in the left temporal area. Lumbar puncture revealed 7 nucleated cells/μl (90 % neutrophils), 295 erythrocytes/μl, and normal glucose and protein. HSV-1 PCR was positive in the CSF and she was treated with aciclovir. One week into her hospitalization, after initially improving she developed right lateral rectus palsy and depressed level of consciousness. Repeat MRI demonstrated increased edema in the left temporal lobe. The patient eventually recovered but she was left with subtle language deficits. Follow-up MRI 6 months after her HSVE demonstrated cystic encephalomalacia in the left anterior temporal lobe.

Magnetic resonance imaging in acute herpes simplex virus-1 encephalitis. (A) Diffusion restriction on diffusion-weighted imaging (DWI) in the left mesial temporal lobe that corresponded to (B, C) fluid-attenuated inversion recovery (FLAIR) hyperintensity. (D) On day 8, with clinical deterioration, there was increased fluid restriction on DWI in the left mesial temporal lobe with tracking along the cortical ribbon that corresponded with (E, F) increased FLAIR hyperintensity and swelling
Edema and Herniation
Cytotoxic and/or vasogenic edema associated with the infectious process or the host immune response can lead to focal or global mass effect and increased intracranial pressure in HSVE. When this is suspected, rapid bedside evaluation and head CT are indicated. Several studies in patients with meningitis have suggested that CT should precede lumbar puncture in patients with signs such as optic disc edema, new seizures, or severe impairment of consciousness (see Table 10 in [65] for a succinct summary of contraindications to lumbar puncture) [134–136]. In practice, we find that most patients have had an initial CT scan in the emergency department prior to neurological evaluation.
Kalanuria et al. [137] recently reviewed the management of herniation. Initial emergency measures that may attenuate intracranial pressure include elevation of the head of the bed to at least 30 degrees, adequate oxygenation with target oxygen saturation > 90 %, and brief (<2 h) hyperventilation with target PaCO2 of 30–35 mmHg. Hyperosmolar therapy with either hypertonic saline or mannitol should be considered in cases where mass effect from significant edema is noted. We favor hypertonic saline over mannitol and, though no randomized clinical trials exist, a meta-analysis has supported this practice [138]. Two percent sodium (Na) solution can be given through a peripheral line, while 3 % or 23.4 % Na should be given through a central line. Boluses of 250–300 ml 2–3 % Na can be given to maintain serum sodium in the range of 150–155, with conversion to maintenance infusion as needed. In active brain herniation, a 30-ml bolus of 23.4 % Na can be given. If the patient is hyponatremic at presentation, sodium must be corrected slowly given the risk of myelin injury, and mannitol may be the safer option. Hypertonic therapy carries risk of myelin injury, subdural hematoma/effusion, rebound cerebral edema, phlebitis, hypotension, pulmonary edema, heart failure, hypokalemia, hyperchloremic acidemia, coagulopathy, and intravascular hemolysis.
In severe cases of cerebral edema refractory to the aforementioned medical management, barbiturate coma and/or decompressive craniectomy should be considered. Case series and case reports suggest the potential for good outcomes, even in cases of bacterial meningitis or viral encephalitis requiring surgical intervention [139]. Patients with evidence of obstructive hydrocephalus should likewise be evaluated for surgical intervention such as external ventricular drainage.
Seizures
Seizures are common in encephalitis and some 15 % of patients have SE during the course of their illness [140–142]. A recently published Cochrane review of the use of antiepileptic medications for the primary and secondary prevention of seizures in viral encephalitis concluded that there was insufficient evidence to support either practice [143]. However, our practice is to provide antiepileptic medications to all patients with seizures and encephalitis given the possibility of excitotoxicity and further brain injury in the setting of recurrent seizures.
Status epilepticus (SE) is defined as seizure lasting > 5 min or recurrent seizure activity without recovery between episodes. A treatment algorithm for the management of patients in SE has recently been published [144], and guidelines for the management of convulsive and nonconvulsive SE are also available [145]. The first priorities of managing patients in SE are airway protection and support of respiration and circulation as needed. Bedside glucose testing should be promptly obtained and hypoglycemia corrected as needed. First-line antiepileptic agents for patients with SE include lorazepam (0.1 mg/kg up to 4 mg per dose given at 5–10-min intervals), midazolam (10 mg intramuscularly), or diazepam (10 mg per rectum). First-line therapy will abort SE in roughly half of all patients [146]. All patients with convulsive SE should be given a second-line agent immediately after administration of the first-line agent in order to prevent further seizures. We prefer valproate sodium (25–40 mg/kg i.v.) [147–149] or fosphenytoin (18–20 phenytoin equivalents/kg i.v.) [150], which are among the best studied antiepileptic therapies in SE. Phenytoin may precipitate hypotension that can generally be corrected by giving a fluid bolus and reducing the rate of infusion. In hemodynamically tenuous patients, we therefore prefer valproate, which can be rapidly infused and is generally well tolerated, even in the critically ill. SE in a patient with HSVE may be a manifestation of increasing edema and mass effect, and emergent brain CT should be considered while treatment is being initiated.
If seizures do not abate with first- and second-line therapy, we initiate anesthetic infusion with propofol or midazolam as our preferred agents, though no one anesthetic has been shown to be superior to the others. This should be titrated to cessation of clinical seizure activity. Continuous EEG should be initiated emergently for patients who are unconscious but without clinical evidence of seizures, as subclinical seizures are common in this setting and can only be diagnosed by EEG. Notably, in patients with subclinical SE, intravenous anesthesia has been associated with increased mortality, suggesting that it should be avoided if possible [151]. Once seizures have been controlled and preventative antiepileptic agents have reached therapeutic doses, infusion is generally maintained for 24 h before controlled taper of anesthetic agents with continuous EEG monitoring.
Among patients who have seizures but do not experience SE, the underlying inflammatory epileptogenic stimulus in HSVE is likely to persist for at least the duration of the illness. Therefore, with the first seizure we begin secondary prevention with an antiepileptic medication such as levetiracetam (starting dose 1000–3000 mg i.v. or p.o.), lacosamide (200–400 mg i.v. or p.o.), valproate sodium (20–40 mg/kg i.v. or p.o.), or other antiepileptic agent, generally based on comorbidities and patient/physician preference. The aforementioned agents can be given with i.v. loading doses.
Case 2: A-79-Year-old Woman in SE
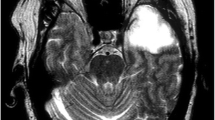
A 79-year-old woman presented in SE 3 months after being treated for HSVE. Brain MRI is shown in Fig. 3. HSV PCR and other infectious studies from the serum and CSF were negative. Anti-NMDAR IgG antibodies were detected in the CSF by immunofluorescence assay at 1:20 (normal: <1:1). With antiepileptic medications, steroids, plasma exchange, and intravenous immunoglobulin (IVIg), the patient improved and was discharged to skilled nursing care. Anti-NMDAR encephalitis and other immune-mediated encephalitides can be triggered by HSV [152]. In contradistinction to the initial HSVE, significant contrast enhancement on brain MRI has been observed [153], and may be a biomarker of autoimmune relapse, though future studies with greater patient numbers are needed. Importantly, these cases of clinical relapse after HSVE appear to respond favorably to immunotherapy [153].

Central nervous system autoimmunity following herpes simplex virus-1 encephalitis. (A–C) Extensive patchy postgadolinium enhancement involving the gray and white matter of the temporal and frontal lobes, and corpus callosum. (D–F) Corresponding fluid-attenuated inversion recovery (FLAIR) sequences demonstrate left > right temporal lobe cystic encephalomalacia and FLAIR hyperintense lesions
Outcomes
HSVE is a cause of significant morbidity and mortality. The mortality of untreated HSV encephalitis is roughly 70 %, and 97 % of survivors will not return to their previous level of function [41, 154–156]. Clinical trials in the 1980s demonstrated significantly improved outcomes with intravenous aciclovir, as described above [103, 104], and the 1-year mortality with current antivirals and supportive care is now in the range of 5–15 %, despite high rates of admission to the ICU [44, 60, 157]. However, consequent neuropsychiatric deficits remain common (69–89 %) [60, 157].
The economic burden of HSVE is also very high. Given the emergence of West Nile virus neuroinvasive disease in the USA and the recognition of immune-mediated etiologies, such as anti-NMDAR encephalitis, Vora et al. [158] estimated the burden of encephalitis-associated hospitalizations from 1998 to 2010, updating a previous study [159]. They reported a mean length of hospital stay of 11.2 days with a median inpatient charge of $48,852 for encephalitis-related hospitalizations and $58,082 for encephalitis related to herpes, in 2010.
Among the most significant negative prognostic factors are older age, coma/lower level of consciousness at presentation, restricted diffusion on DWI, and delay in aciclovir administration [57]. Sutter et al. [90] observed that a normal EEG was the independent factor most strongly associated with survival. Kim et al. [160] recently retrospectively reviewed 29 patients with PCR-proven HSVE and found that severe EEG abnormalities were predictive of poor outcome at 6 months, although this was not observed in a series of 45 patients from the Mayo Clinic [57]. Early recognition and timely administration of aciclovir are critically important for improving outcomes, and late administration of aciclovir is the most readily modifiable risk factor for poor outcomes [62, 63, 161, 162]. Factors contributing to delayed treatment include immunocompromise [63], severe comorbid disease, history of alcohol abuse, absence of fever, and CSF leukocytes <10/ml [162].
Immune-Mediated Encephalitis and Apparent HSVE Relapse
While most cases of HSVE are monophasic, a subset of patients return to medical attention with an apparent clinical relapse after completing treatment. Most are children who present with choreoathetosis [163]; however, patients of all ages may present with a variety of neurologic manifestations such as new changes in behavior or personality, memory deficits, and seizures. The frequency of clinical relapse has been reported to range from 5 % to 27 %, with the higher frequencies observed in children [163–166]. The relapse is generally less severe than the initial illness; however, fatal cases have been reported [167].
While viral relapse is possible and some cases have had at least transient HSV PCR positivity during the relapse episode [165], many have no evidence of HSV activity, as demonstrated by negative HSV PCR from the CSF and poor clinical response to antiviral medications. An immune-mediated process has long been suspected in this setting. In one study, 32 consecutive adults with CSF PCR- or serology-proven HSVE who were treated with aciclovir or vidarabine were prospectively followed for relapse, which occurred in 4 patients [168]. However, none of these had HSV PCR positivity in the CSF during the apparent relapse, and markers of neural and glial cell damage (including neuron-specific enolase, S-100, and glial fibrillary acidic protein) were markedly lower in the CSF during relapse than on initial presentation. The authors concluded that direct viral cytotoxicity was not the mechanism of relapse, but rather suggested an immune-mediated process.
Recent evidence has supported the immune-mediated hypothesis. Multiple case reports and, more recently, case series have demonstrated that many patients with HSVE relapse develop anti-NMDAR immunoglobulins [152, 169–174]. Antibodies targeting other known neuronal antigens and unidentified neuronal antigens have also been reported in this setting [152, 153, 174, 175]. The precise pathogenic mechanisms remain to be elucidated, but may involve mechanisms of molecular mimicry or an autoimmune response to the release of neuronal antigens associated with host cell lysis.
Armangue et al. [153] recently reported 14 patients with HSVE relapse and compared the clinical, imaging, and laboratory features in adults and teenagers (median age 40 years, range 13–69 years) with those in young children (median age 13 months, range 6–20 months). Older patients were significantly less likely to have choreoathetosis or decreased level of consciousness compared with children. Moreover, diagnosis and treatment were delayed in older patients [85 days from relapse symptom onset to antibody testing (range 17–296 days) vs 4 days (range 0–55 days) in children; p = 0.037]. In some cases, the development of neuronal autoantibodies may occur early in the illness and the syndrome can appear as progression of the initial HSVE episode. Brain MRI during the relapse episode frequently demonstrated contrast enhancement that improved with immunomodulatory therapy.
Patients with post-HSVE immune-mediated encephalitis are likely to respond favorably to immunotherapy but may be left with neurological deficits attributable to the HSVE. First-line treatment with steroids and/or IVIg or plasma exchange resulted in substantial improvement in all patients in the series by Armangue et al. [153]. One patient who had SE transiently improved with plasma exchange but developed further seizures and required second-line treatment with rituximab and IVIg.
Given the recent data outlined above, clinicians should be aware of the risk of immune-mediated relapse after HSVE, which is likely an under-recognized complication. Early follow-up (i.e., within 1 month of discharge) should be strongly considered in order to monitor for evidence of immune-mediated complications, which may be misdiagnosed as progression or recrudescence of HVSE deficits. A high index of suspicion is warranted, particularly in adults, who are less likely to present with stereotyped neurologic manifestations such as chorea. Evaluation for viral relapse with HSV PCR from the CSF should be coupled with evaluation of an immune-mediated etiology by historical and examination findings and testing for autoantibodies from the CSF. If clinical suspicion is high, sending specimens to a research laboratory with expertise in immune-mediated encephalitides should be considered and evaluation for antibodies targeting unidentified neuronal antigens may be fruitful. Brain MRI is helpful in identifying other possible post-HSVE complications that might mimic relapse and contrast enhancement may be a marker of immune-mediated sequelae [153]. Once viral reactivation or persistence have been excluded, treatment with immunomodulatory therapy should be strongly considered with a combination of steroids and IVIg as a reasonable first-line regimen. Second-line therapy with rituximab and/or cyclophosphamide is reasonable in patients who do not respond to first-line agents. Future studies investigating the epidemiology, pathophysiology, and optimal clinical management of these patients are warranted.
Future Lines of Investigation
Although significant advances in the treatment of HSVE have been made since the first reports in the 1940s, there is still a great need to improve outcomes. The diagnostic challenges presented by encephalitis and the high frequency of idiopathic cases stresses the importance of improving our diagnostic approach to the encephalitic patient. Further studies are needed in order to determine what contribution the host immune system plays in damaging the CNS, and mechanisms of such damage remain to be fully elucidated. Deepening our understanding of the role of host immunity in HSV pathogenicity may have significant implications for attenuating the long-term sequelae of HSVE and further investigations in this area should be pursued. Methods capable of decreasing the long-term neurocognitive deficits in patients with HSVE are also greatly needed. The development of a vaccine to prevent primary infection with HSV is an area of active research and has the potential to prevent serious complications of HSV infection [176].
References
Venkatesan A, Tunkel AR, Bloch KC, et al. Case definitions, diagnostic algorithms, and priorities in encephalitis: consensus statement of the international encephalitis consortium. Clin Infect Dis 2013;57:1114-1128.
Venkatesan A. Epidemiology and outcomes of acute encephalitis. Curr Opin Neurol 2015;28:277-282.
Goodpasture EW. Herpetic infection, with especial reference to involvement of the nervous system. 1929. Medicine 1993;72:125-132.
Commission M. Epidemic encephalitis: etiology, epidemiology, treatment. Report of a survey by the Mathewson Commission. New York: Columbia university Press; 1929.
Smith MG, Lennette EH, Reames HR. Isolation of the virus of herpes simplex and the demonstration of intranuclear inclusions in a case of acute encephalitis. Am J Pathol 1941;17:55-68.
Zarafonetis CJ, Smadel JE. Fatal herpes simplex encephalitis in man. Am J Pathol 1944;20:429-445.
Shieh MT, Spear PG. Herpesvirus-induced cell fusion that is dependent on cell surface heparan sulfate or soluble heparin. J Virol 1994;68:1224-1228.
Mata M, Zhang M, Hu X, Fink DJ. HveC (nectin-1) is expressed at high levels in sensory neurons, but not in motor neurons, of the rat peripheral nervous system. J Neurovirol 2001;7:476-480.
Shukla ND, Tiwari V, Valyi-Nagy T. Nectin-1-specific entry of herpes simplex virus 1 is sufficient for infection of the cornea and viral spread to the trigeminal ganglia. Mol Vis 2012;18:2711-2716.
Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL. Transport and egress of herpes simplex virus in neurons. Rev Med Virol 2008;18:35-51.
Smith G. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 2012;66:153-176.
Mori I, Nishiyama Y, Yokochi T, Kimura Y. Olfactory transmission of neurotropic viruses. J Neurovirol 2005;11:129-137.
Jennische E, Eriksson CE, Lange S, Trybala E, Bergstrom T. The anterior commissure is a pathway for contralateral spread of herpes simplex virus type 1 after olfactory tract infection. J Neurovirol 2015;21:129-147.
Dinn JJ. Transolfactory spread of virus in herpes simplex encephalitis. Br Med J 1980;281:1392.
Ojeda VJ, Archer M, Robertson TA, Bucens MR. Necropsy study of the olfactory portal of entry in herpes simplex encephalitis. Med J Aust 1983;1:79-81.
Twomey JA, Barker CM, Robinson G, Howell DA. Olfactory mucosa in herpes simplex encephalitis. J Neurol Neurosurg Psychiatry 1979;42:983-987.
Davis LE, Johnson RT. An explanation for the localization of herpes simplex encephalitis? Ann Neurol 1979;5:2-5.
Tyler KL, Tedder DG, Yamamoto LJ, et al. Recurrent brainstem encephalitis associated with herpes simplex virus type 1 DNA in cerebrospinal fluid. Neurology 1995;45:2246-2250.
Rose JW, Stroop WG, Matsuo F, Henkel J. Atypical herpes simplex encephalitis: clinical, virologic, and neuropathologic evaluation. Neurology 1992;42:1809-1812.
Hamilton RL, Achim C, Grafe MR, Fremont JC, Miners D, Wiley CA. Herpes simplex virus brainstem encephalitis in an AIDS patient. Clin Neuropathol 1995;14:45-50.
Steiner I, Spivack JG, O'Boyle DR, 2nd, Lavi E, Fraser NW. Latent herpes simplex virus type 1 transcription in human trigeminal ganglia. J Virol 1988;62:3493-3496.
Steiner I. Herpes simplex virus encephalitis: new infection or reactivation? Curr Opin Neurol 2011;24:268-274.
Fraser NW, Lawrence WC, Wroblewska Z, Gilden DH, Koprowski H. Herpes simplex type 1 DNA in human brain tissue. Proc Natl Acad Sci U S A 1981;78:6461-6465.
Whitley R, Lakeman AD, Nahmias A, Roizman B. Dna restriction-enzyme analysis of herpes simplex virus isolates obtained from patients with encephalitis. N Engl J Med 1982;307:1060-1062.
Medzhitov R, Janeway CA, Jr. Decoding the patterns of self and nonself by the innate immune system. Science 2002;296:298-300.
Zhang SY, Jouanguy E, Sancho-Shimizu V, et al. Human Toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol Rev 2007;220:225-236.
Malmgaard L. Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res 2004;24:439-454.
Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev 2001;14:778-809.
Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007;317:1522-1527.
Zhang SY, Casanova JL. Inborn errors underlying herpes simplex encephalitis: From TLR3 to IRF3. J Exp Med 2015;212:1342-1343.
Andersen LL, Mork N, Reinert LS, et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med 2015;212:1371-1379.
Lundberg P, Ramakrishna C, Brown J, et al. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol 2008;82:7078-7088.
Marques CP, Cheeran MC, Palmquist JM, Hu S, Urban SL, Lokensgard JR. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J Immunol 2008;181:6417-6426.
Rock DL, Fraser NW. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature 1983;302:523-525.
Oldstone MB. Molecular anatomy of viral persistence. J Virol 1991;65:6381-6386.
Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 2008;6:211-221.
Egan KP, Wu S, Wigdahl B, Jennings SR. Immunological control of herpes simplex virus infections. J Neurovirol 2013;19:328-345.
Roizman B, Zhou G, Du T. Checkpoints in productive and latent infections with herpes simplex virus 1: conceptualization of the issues. J Neurovirol 2011;17:512-517.
Smith JS, Robinson NJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis 2002;186(Suppl. 1):S3-S28.
Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2--United States, 1999-2010. J Infect Dis 2014;209:325-333.
Steiner I, Benninger F. Update on herpes virus infections of the nervous system. Curr Neurol Neurosci Rep 2013;13:414.
Granerod J, Ambrose HE, Davies NW, et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010;10:835-844.
Tunkel AR, Glaser CA, Bloch KC, et al. The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis 2008;47:303-327.
Hjalmarsson A, Blomqvist P, Skoldenberg B. Herpes simplex encephalitis in Sweden, 1990-2001: incidence, morbidity, and mortality. Clin Infect Dis 2007;45:875-880.
Mailles A, Stahl JP, Steering C, Investigators G. Infectious encephalitis in france in 2007: a national prospective study. Clin Infect Dis 2009;49:1838-1847.
Glaser CA, Gilliam S, Schnurr D, et al. In search of encephalitis etiologies: diagnostic challenges in the California Encephalitis Project, 1998–2000. Clin Infect Dis 2003;36:731-742.
Dagsdottir HM, Sigurethardottir B, Gottfreethsson M, et al. Herpes simplex encephalitis in Iceland 1987–2011. Springerplus 2014;3:524.
de Ory F, Avellon A, Echevarria JE, et al. Viral infections of the central nervous system in Spain: a prospective study. J Med Virol 2013;85:554-562.
Quist-Paulsen E, Kran AM, Dunlop O, Wilson J, Ormaasen V. Infectious encephalitis: a description of a Norwegian cohort. Scand J Infect Dis 2013;45:179-185.
Child N, Croxson MC, Rahnama F, Anderson NE. A retrospective review of acute encephalitis in adults in Auckland over a five-year period (2005–2009). J Clin Neurosci 2012;19:1483-1485.
Choi R, Kim GM, Jo IJ, et al. Incidence and clinical features of herpes simplex viruses (1 and 2) and varicella-zoster virus infections in an adult Korean population with aseptic meningitis or encephalitis. J Med Virol 2014;86:957-962.
Barbadoro P, Marigliano A, Ricciardi A, D'Errico MM, Prospero E. Trend of hospital utilization for encephalitis. Epidemiol Infect 2012;140:753-764.
Scheld WM, Whitley RJ, Marra CM. Infections of the central nervous system. 4th ed. Philadelphia: Wolters Kluwer Health; 2014. 907 p.
Abel L, Plancoulaine S, Jouanguy E, et al. Age-dependent Mendelian predisposition to herpes simplex virus type 1 encephalitis in childhood. J Pediatr 2010;157:623-629, 9 e1.
Whitley RJ. Viral encephalitis. N Engl J Med 1990;323:242-250.
George BP, Schneider EB, Venkatesan A. Encephalitis hospitalization rates and inpatient mortality in the United States, 2000–2010. PLOS ONE 2014;9:e104169.
Singh TD, Fugate JE, Hocker S, Wijdicks EF, Aksamit AJ, Jr., Rabinstein AA. Predictors of outcome in HSV encephalitis. J Neurol 2016;263:277-289.
Riancho J, Delgado-Alvarado M, Sedano MJ, Polo JM, Berciano J. Herpes simplex encephalitis: clinical presentation, neurological sequelae and new prognostic factors. Ten years of experience. Neurol Sci 2013;34:1879-1881.
Gilden DH, Mahalingam R, Cohrs RJ, Tyler KL. Herpesvirus infections of the nervous system. Nat Clin Pract Neurol 2007;3:82-94.
Sili U, Kaya A, Mert A, Group HSVES. Herpes simplex virus encephalitis: clinical manifestations, diagnosis and outcome in 106 adult patients. J Clin Virol 2014;60:112-118.
Riera-Mestre A, Gubieras L, Martinez-Yelamos S, Cabellos C, Fernandez-Viladrich P. Adult herpes simplex encephalitis: fifteen years' experience. Enferm Infecc Microbiol Clin 2009;27:143-147.
Raschilas F, Wolff M, Delatour F, et al. Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a multicenter study. Clin Infect Dis 2002;35:254-260.
Tan IL, McArthur JC, Venkatesan A, Nath A. Atypical manifestations and poor outcome of herpes simplex encephalitis in the immunocompromised. Neurology 2012;79:2125-2132.
Schiff D, Rosenblum MK. Herpes simplex encephalitis (HSE) and the immunocompromised: a clinical and autopsy study of HSE in the settings of cancer and human immunodeficiency virus-type 1 infection. Hum Pathol 1998;29:215-222.
Solomon T, Michael BD, Smith PE, et al. Management of suspected viral encephalitis in adults—Association of British Neurologists and British Infection Association National Guidelines. J Infect 2012;64:347-373.
Cesario TC, Poland JD, Wulff H, Chin TD, Wenner HA. Six years experience with herpes simplex virus in a children's home. Am J Epidemiol 1969;90:416-422.
Steiner I, Schmutzhard E, Sellner J, Chaudhuri A, Kennedy PG, European Federation of Neurological S, et al. EFNS-ENS guidelines for the use of PCR technology for the diagnosis of infections of the nervous system. Eur J Neurol 2012;19:1278-1291.
Lakeman FD, Whitley RJ. Diagnosis of herpes simplex encephalitis: application of polymerase chain reaction to cerebrospinal fluid from brain-biopsied patients and correlation with disease. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J Infect Dis 1995;171:857-863.
Behzad-Behbahani A, Abdolvahab A, Gholamali YP, Roshanak B, Mahmood R. Clinical signs as a guide for performing HSV-PCR in correct diagnosis of herpes simplex virus encephalitis. Neurol India 2003;51:341-344.
Weisberg LA, Greenberg J, Stazio A. Computed tomographic findings in acute viral encephalitis in adults with emphasis on herpes simplex encephalitis. Comput Med Imaging Graph 1988;12:385-392.
Leo JS, Weiner RL, Lin JP, Ransohoff J. Computed tomography in herpes simplex encephalitis. Surg Neurol 1978;10:313-317.
Hindmarsh T, Lindqvist M, Olding-Stenkvist E, Skoldenberg B, Forsgren M. Accuracy of computed tomography in the diagnosis of herpes simplex encephalitis. Acta Radiol Suppl 1986;369:192-196.
Schroth G, Gawehn J, Thron A, Vallbracht A, Voigt K. Early diagnosis of herpes simplex encephalitis by MRI. Neurology 1987;37:179-183.
Domingues RB, Fink MC, Tsanaclis AM, et al. Diagnosis of herpes simplex encephalitis by magnetic resonance imaging and polymerase chain reaction assay of cerebrospinal fluid. J Neurol Sci 1998;157:148-153.
Misra UK, Kalita J, Phadke RV, et al. Usefulness of various MRI sequences in the diagnosis of viral encephalitis. Acta Trop 2010;116:206-211.
Sawlani V. Diffusion-weighted imaging and apparent diffusion coefficient evaluation of herpes simplex encephalitis and Japanese encephalitis. J Neurol Sci 2009;287:221-226.
McCabe K, Tyler K, Tanabe J. Diffusion-weighted MRI abnormalities as a clue to the diagnosis of herpes simplex encephalitis. Neurology 2003;61:1015-1016.
Kuker W, Nagele T, Schmidt F, Heckl S, Herrlinger U. Diffusion-weighted MRI in herpes simplex encephalitis: a report of three cases. Neuroradiology 2004;46:122-125.
Obeid M, Franklin J, Shrestha S, Johnson L, Quattromani F, Hurst D. Diffusion-weighted imaging findings on MRI as the sole radiographic findings in a child with proven herpes simplex encephalitis. Pediatr Radiol 2007;37:1159-1162.
Okanishi T, Yamamoto H, Hosokawa T, et al. Diffusion-weighted MRI for early diagnosis of neonatal herpes simplex encephalitis. Brain Develop 2015;37:423-431.
Dhawan A, Kecskes Z, Jyoti R, Kent AL. Early diffusion-weighted magnetic resonance imaging findings in neonatal herpes encephalitis. J Paediatr Child Health 2006;42:824-826.
Duckworth JL, Hawley JS, Riedy G, Landau ME. Magnetic resonance restricted diffusion resolution correlates with clinical improvement and response to treatment in herpes simplex encephalitis. Neurocrit Care 2005;3:251-253.
Renard D, Nerrant E, Lechiche C. DWI and FLAIR imaging in herpes simplex encephalitis: a comparative and topographical analysis. J Neurol 2015;262:2101-2105.
Chow FC, Glaser CA, Sheriff H, et al. Use of clinical and neuroimaging characteristics to distinguish temporal lobe herpes simplex encephalitis from its mimics. Clin Infect Dis 2015;60:1377-1383.
Lai CW, Gragasin ME. Electroencephalography in herpes simplex encephalitis. J Clin Neurophysiol 1988;5:87-103.
Verma R, Raut TP, Giri P, Praharaj HN. New onset refractory status epilepticus (NORSE) as the heralding manifestation of herpes simplex encephalitis. BMJ Case Rep 2013;2013.
Sellner J, Trinka E. Seizures and epilepsy in herpes simplex virus encephalitis: current concepts and future directions of pathogenesis and management. J Neurol 2012;259:2019-2030.
Brodtkorb E, Lindqvist M, Jonsson M, Gustafsson A. Diagnosis of herpes simplex encephalitis. A comparison between electroencephalography and computed tomography findings. Acta Neurol Scand 1982;66:462-471.
Upton A, Gumpert J. Electroencephalography in diagnosis of herpes-simplex encephalitis. Lancet 1970;1:650-652.
Sutter R, Kaplan PW, Cervenka MC, et al. Electroencephalography for diagnosis and prognosis of acute encephalitis. Clin Neurophysiol 2015;126:1524-1531.
Ch'ien LT, Boehm RM, Robinson H, Liu C, Frenkel LD. Characteristic early electroencephalographic changes in herpes simplex encephalitis. Arch Neurol 1977;34:361-364.
Illis LS, Taylor FM. The electroencephalogram in herpes-simplex encephalitis. Lancet 1972;1:718-721.
Whitley RJ, Cobbs CG, Alford CA, Jr., et al. Diseases that mimic herpes simplex encephalitis. Diagnosis, presentation, and outcome. NIAD Collaborative Antiviral Study Group. JAMA 1989;262:234-239.
Kaewpoowat Q, Salazar L, Aguilera E, Wootton SH, Hasbun R. Herpes simplex and varicella zoster CNS infections: clinical presentations, treatments and outcomes. Infection 2015 Dec 17 [Epub ahead of print].
Whitley RJ, Soong SJ, Linneman C, Jr., Liu C, Pazin G, Alford CA. Herpes simplex encephalitis. Clinical Assessment. JAMA 1982;247:317-320.
Baringer JR, Pisani P. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol 1994;36:823-829.
Sanders VJ, Waddell AE, Felisan SL, Li X, Conrad AJ, Tourtellotte WW. Herpes simplex virus in postmortem multiple sclerosis brain tissue. Arch Neurol 1996;53:125-133.
Weil AA, Glaser CA, Amad Z, Forghani B. Patients with suspected herpes simplex encephalitis: rethinking an initial negative polymerase chain reaction result. Clin Infect Dis 2002;34:1154-1157.
Elbers JM, Bitnun A, Richardson SE, et al. A 12-year prospective study of childhood herpes simplex encephalitis: is there a broader spectrum of disease? Pediatrics 2007;119:e399-e407.
De Tiege X, Heron B, Lebon P, Ponsot G, Rozenberg F. Limits of early diagnosis of herpes simplex encephalitis in children: a retrospective study of 38 cases. Clin Infect Dis 2003;36:1335-1339.
Venkatesan A, Geocadin RG. Diagnosis and management of acute encephalitis: A practical approach. Neurol Clin Pract 2014;4:206-215.
Eidelman LA, Putterman D, Putterman C, Sprung CL. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA 1996;275:470-473.
Whitley RJ, Alford CA, Hirsch MS, et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med 1986;314:144-149.
Skoldenberg B, Forsgren M, Alestig K, et al. Acyclovir versus vidarabine in herpes simplex encephalitis. Randomised multicentre study in consecutive Swedish patients. Lancet 1984;2:707-711.
Whitley RJ, Gnann JW, Jr. Acyclovir: a decade later. N Engl J Med 1992;327:782-789.
Hirsch MS, Swartz MN. Drug therapy: antiviral agents (second of two parts). N Engl J Med 1980;302:949-953.
Heintz BH, Matzke GR, Dager WE. Antimicrobial dosing concepts and recommendations for critically ill adult patients receiving continuous renal replacement therapy or intermittent hemodialysis. Pharmacotherapy 2009;29:562-577.
de Miranda P, Blum MR. Pharmacokinetics of acyclovir after intravenous and oral administration. J Antimicrob Chemother 1983;12(Suppl. B):29-37.
Sawyer MH, Webb DE, Balow JE, Straus SE. Acyclovir-induced renal failure. Clinical course and histology. Am J Med 1988;84:1067-1071.
Pacheco LR, Tavares HM, Moyses Neto M, et al. [Acute renal failure related to intravenous acyclovir]. Rev Assoc Med Bras 2005;51:275-278.
Adair JC, Gold M, Bond RE. Acyclovir neurotoxicity: clinical experience and review of the literature. South Med J 1994;87:1227-1231.
Pasternak B, Hviid A. Use of acyclovir, valacyclovir, and famciclovir in the first trimester of pregnancy and the risk of birth defects. JAMA 2010;304:859-866.
Rozenberg F, Deback C, Agut H. Herpes simplex encephalitis : from virus to therapy. Infect Disord Drug Targets 2011;11:235-250.
Chatis PA, Crumpacker CS. Resistance of herpesviruses to antiviral drugs. Antimicrob Agents Chemother 1992;36:1589-1595.
Levin MJ, Bacon TH, Leary JJ. Resistance of herpes simplex virus infections to nucleoside analogues in HIV-infected patients. Clin Infect Dis 2004;39(Suppl. 5):S248-S257.
Erlich KS, Mills J, Chatis P, et al. Acyclovir-resistant herpes simplex virus infections in patients with the acquired immunodeficiency syndrome. N Engl J Med 1989;320:293-296.
Hardy WD. Foscarnet treatment of acyclovir-resistant herpes simplex virus infection in patients with acquired immunodeficiency syndrome: preliminary results of a controlled, randomized, regimen-comparative trial. Am J Med 1992;92:30S-35S.
Losada I, Canizares A, Hellin T, Marti-Belda P, Guerrero A. [In vitro susceptibility study of herpes simplex virus to acyclovir and foscarnet. Are routine susceptibility studies necessary?]. Enferm Infecc Microbiol Clin 2002;20:25-27.
Superti F, Ammendolia MG, Marchetti M. New advances in anti-HSV chemotherapy. Curr Med Chem 2008;15:900-911.
Wagstaff AJ, Bryson HM. Foscarnet. A reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs 1994;48:199-226.
Trifillis AL, Cui X, Drusano GL. Use of human renal proximal tubule cell cultures for studying foscarnet-induced nephrotoxicity in vitro. Antimicrob Agents Chemother 1993;37:2496-2499.
Markham A, Faulds D. Ganciclovir. An update of its therapeutic use in cytomegalovirus infection. Drugs 1994;48:455-484.
VanLandingham KE, Marsteller HB, Ross GW, Hayden FG. Relapse of herpes simplex encephalitis after conventional acyclovir therapy. JAMA 1988;259:1051-1053.
Dennett C, Klapper PE, Cleator GM. Polymerase chain reaction in the investigation of "relapse" following herpes simplex encephalitis. J Med Virol 1996;48:129-132.
Cinque P, Cleator GM, Weber T, Monteyne P, Sindic CJ, van Loon AM. The role of laboratory investigation in the diagnosis and management of patients with suspected herpes simplex encephalitis: a consensus report. The EU Concerted Action on Virus Meningitis and Encephalitis. J Neurol Neurosurg Psychiatry 1996;61:339-345.
Solomon T, Hart IJ, Beeching NJ. Viral encephalitis: a clinician's guide. Pract Neurol 2007;7:288-305.
Gnann JW, Jr., Skoldenberg B, Hart J, et al. Herpes simplex encephalitis: lack of clinical benefit of long-term valacyclovir therapy. Clin Infect Dis 2015;61:683-691.
Meyding-Lamade UK, Oberlinner C, Rau PR, et al. Experimental herpes simplex virus encephalitis: a combination therapy of acyclovir and glucocorticoids reduces long-term magnetic resonance imaging abnormalities. J Neurovirol 2003;9:118-125.
Habel AH, Brown JK. Dexamethasone in herpes-simplex encephalitis. Lancet 1972;1:695.
Upton AR, Foster JB, Barwick DD. Dexamethasone treatment in herpes-simplex encephalitis. Lancet 1971;1:861.
Kamei S, Sekizawa T, Shiota H, et al. Evaluation of combination therapy using aciclovir and corticosteroid in adult patients with herpes simplex virus encephalitis. J Neurol Neurosurg Psychiatry 2005;76:1544-1549.
Martinez-Torres F, Menon S, Pritsch M, et al. Protocol for German trial of Acyclovir and corticosteroids in Herpes-simplex-virus-encephalitis (GACHE): a multicenter, multinational, randomized, double-blind, placebo-controlled German, Austrian and Dutch trial [ISRCTN45122933]. BMC Neurol 2008;8:40.
Conrady CD, Drevets DA, Carr DJ. Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J Neuroimmunol 2010;220:1-9.
van Crevel H, Hijdra A, de Gans J. Lumbar puncture and the risk of herniation: when should we first perform CT? J Neurol 2002;249:129-137.
Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med 2001;345:1727-1733.
van de Beek D, de Gans J, Spanjaard L, Weisfelt M, Reitsma JB, Vermeulen M. Clinical features and prognostic factors in adults with bacterial meningitis. N Engl J Med 2004;351:1849-1859.
Kalanuria AA, Geocadin RG, Puttgen HA. Brain code and coma recovery: aggressive management of cerebral herniation. Semin Neurol 2013;33:133-141.
Kamel H, Navi BB, Nakagawa K, Hemphill JC, 3rd, Ko NU. Hypertonic saline versus mannitol for the treatment of elevated intracranial pressure: a meta-analysis of randomized clinical trials. Crit Care Med 2011;39:554-559.
Safain MG, Roguski M, Kryzanski JT, Weller SJ. A review of the combined medical and surgical management in patients with herpes simplex encephalitis. Clin Neurol Neurosurg 2015;128:10-16.
Thakur KT, Motta M, Asemota AO, et al. Predictors of outcome in acute encephalitis. Neurology 2013;81:793-800.
Misra UK, Tan CT, Kalita J. Viral encephalitis and epilepsy. Epilepsia 2008;49(Suppl. 6):13-18.
Kalita J, Nair PP, Misra UK. Status epilepticus in encephalitis: a study of clinical findings, magnetic resonance imaging, and response to antiepileptic drugs. J Neurovirol 2008;14:412-417.
Pandey S, Rathore C, Michael BD. Antiepileptic drugs for the primary and secondary prevention of seizures in viral encephalitis. Cochrane Database Syst Rev 2014;10:CD010247.
Hocker SE. Status epilepticus. Continuum 2015;21(5 Neurocritical Care):1362-1383.
Brophy GM, Bell R, Claassen J, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012;17:3-23.
Alldredge BK, Gelb AM, Isaacs SM, et al. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med 2001;345:631-637.
Misra UK, Kalita J, Patel R. Sodium valproate vs phenytoin in status epilepticus: a pilot study. Neurology 2006;67:340-342.
Agarwal P, Kumar N, Chandra R, Gupta G, Antony AR, Garg N. Randomized study of intravenous valproate and phenytoin in status epilepticus. Seizure 2007;16:527-532.
Trinka E, Hofler J, Zerbs A, Brigo F. Efficacy and safety of intravenous valproate for status epilepticus: a systematic review. CNS Drugs 2014;28:623-639.
Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med 1998;339:792-798.
Sutter R, Marsch S, Fuhr P, Kaplan PW, Ruegg S. Anesthetic drugs in status epilepticus: risk or rescue? A 6-year cohort study. Neurology 2014;82:656-664.
Armangue T, Leypoldt F, Malaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014;75:317-323.
Armangue T, Moris G, Cantarin-Extremera V, et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology 2015;85:1736-1743.
Tyler KL. Herpes simplex virus infections of the central nervous system: encephalitis and meningitis, including Mollaret's. Herpes 2004;11(Suppl. 2):57A-64A.
Kimberlin DW. Management of HSV encephalitis in adults and neonates: diagnosis, prognosis and treatment. Herpes 2007;14:11-16.
Whitley RJ. Herpes simplex encephalitis: adolescents and adults. Antiviral Res 2006;71:141-148.
Stahl JP, Mailles A, De Broucker T, Steering C, Investigators Group. Herpes simplex encephalitis and management of acyclovir in encephalitis patients in France. Epidemiol Infect 2012;140:372-381.
Vora NM, Holman RC, Mehal JM, Steiner CA, Blanton J, Sejvar J. Burden of encephalitis-associated hospitalizations in the United States, 1998-2010. Neurology 2014;82:443-451.
Khetsuriani N, Holman RC, Anderson LJ. Burden of encephalitis-associated hospitalizations in the United States, 1988-1997. Clin Infect Dis 2002;35:175-182.
Kim YS, Jung KH, Lee ST, et al. Prognostic value of initial standard EEG and MRI in patients with herpes simplex encephalitis. J Clin Neurol 2016;12:224-229.
McGrath N, Anderson NE, Croxson MC, Powell KF. Herpes simplex encephalitis treated with acyclovir: diagnosis and long term outcome. J Neurol Neurosurg Psychiatry 1997;63:321-326.
Poissy J, Wolff M, Dewilde A, et al. Factors associated with delay to acyclovir administration in 184 patients with herpes simplex virus encephalitis. Clin Microbiol Infect 2009;15:560-564.
De Tiege X, Rozenberg F, Des Portes V, et al. Herpes simplex encephalitis relapses in children: differentiation of two neurologic entities. Neurology 2003;61:241-243.
Barthez-Carpentier MA, Rozenberg F, Dussaix E, et al. Relapse of herpes simplex encephalitis. J Child Neurol 1995;10:363-368.
Ito Y, Kimura H, Yabuta Y, et al. Exacerbation of herpes simplex encephalitis after successful treatment with acyclovir. Clin Infect Dis 2000;30:185-187.
Schleede L, Bueter W, Baumgartner-Sigl S, et al. Pediatric herpes simplex virus encephalitis: a retrospective multicenter experience. J Child Neurol 2013;28:321-331.
Barthez MA, Billard C, Santini JJ, Ruchoux MM, Grangeponte MC. Relapse of herpes simplex encephalitis. Neuropediatrics 1987;18:3-7.
Skoldenberg B, Aurelius E, Hjalmarsson A, et al. Incidence and pathogenesis of clinical relapse after herpes simplex encephalitis in adults. J Neurol 2006;253:163-170.
Armangue T, Titulaer MJ, Malaga I, et al. Pediatric anti-N-methyl-D-aspartate receptor encephalitis-clinical analysis and novel findings in a series of 20 patients. J Pediatr 2013;162:850-856 e2.
Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus-1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013;81:1637-1639.
Bektas O, Tanyel T, Kocabas BA, Fitoz S, Ince E, Deda G. Anti-N-methyl-D-aspartate receptor encephalitis that developed after herpes encephalitis: a case report and literature review. Neuropediatrics 2014;45:396-401.
Pruss H, Finke C, Holtje M, et al. N-methyl-D-aspartate receptor antibodies in herpes simplex encephalitis. Ann Neurol 2012;72:902-911.
Hacohen Y, Deiva K, Pettingill P, et al. N-methyl-D-aspartate receptor antibodies in post-herpes simplex virus encephalitis neurological relapse. Mov Disord 2014;29:90-96.
Mohammad SS, Sinclair K, Pillai S, et al. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to N-Methyl-D-aspartate receptor or dopamine-2 receptor. Mov Disord 2014;29:117-122.
Bradshaw MJ, Pawate S, Lennon VA, Bloch KC, Brown KM. Herpes simplex virus 1 encephalitis associated with voltage-gated calcium channel autoimmunity. Neurology 2015;85:2176-2177.
Johnston C, et al. Status of vaccine research and development of vaccines for herpes simplex virus prepared for WHO PD-VAC. Vaccine 2016 Mar 11 [Epub ahead of print].
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Bradshaw, M.J., Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics 13, 493–508 (2016). https://doi.org/10.1007/s13311-016-0433-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0433-7