
Abstract
Palmitoylethanolamide (PEA) is an endogenous lipid mediator known to reduce pain and inflammation. However, only limited clinical studies have evaluated the effects of PEA in neuroinflammatory and neurodegenerative diseases. Multiple sclerosis (MS) is a chronic autoimmune and inflammatory disease of the central nervous system. Although subcutaneous administration of interferon (IFN)-β1a is approved as first-line therapy for the treatment of relapsing–remitting MS (RR-MS), its commonly reported adverse events (AEs) such as pain, myalgia, and erythema at the injection site, deeply affect the quality of life (QoL) of patients with MS. In this randomized, double-blind, placebo-controlled study, we tested the effect of ultramicronized PEA (um-PEA) added to IFN-β1a in the treatment of clinically defined RR-MS. The primary objectives were to estimate whether, with um-PEA treatment, patients with MS perceived an improvement in pain and a decrease of the erythema width at the IFN-β1a injection site in addition to an improvement in their QoL. The secondary objectives were to evaluate the effects of um-PEA on circulating interferon-γ, tumor necrosis factor-α, and interleukin-17 serum levels, N-acylethanolamine plasma levels, Expanded Disability Status Scale (EDSS) progression, and safety and tolerability after 1 year of treatment. Patients with MS receiving um-PEA perceived an improvement in pain sensation without a reduction of the erythema at the injection site. A significant improvement in QoL was observed. No significant difference was reported in EDSS score, and um-PEA was well tolerated. We found a significant increase of palmitoylethanolamide, anandamide and oleoylethanolamide plasma levels, and a significant reduction of interferon-γ, tumor necrosis factor-α, and interleukin-17 serum profile compared with the placebo group. Our results suggest that um-PEA may be considered as an appropriate add-on therapy for the treatment of IFN-β1a-related adverse effects in RR-MS.
Similar content being viewed by others

Introduction
Palmitoylethanolamide (PEA) is an endogenous lipid mediator and a member of the N-acylethanolamine (NAE) family of bioactive lipids. PEA is present in the central nervous system (CNS) [1]; however, its exact role in the CNS during neuroinflammatory processes remains to be fully understood. Since the 1990s, interest in the therapeutic potential of PEA has increased owing to the discovery of the effects of PEA in many preclinical paradigms for pain and chronic inflammation [2]. PEA has antinociceptive properties in several animal models [3–5], prevents neurotoxicity and neurodegeneration [6–8], and inhibits peripheral inflammation and mast cell degranulation [9, 10]. These effects of PEA are not only observed when used as a drug (i.e., after direct administration) [11, 12], but also when its endogenous levels are increased by blocking its catabolism [13]. Although the receptors mediating the effects of PEA are not fully characterized, its anti-inflammatory effects have been associated with peroxisome proliferator-activated receptor-α (PPAR-α) activation [11, 14, 15].
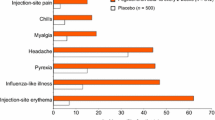
Multiple sclerosis (MS) is a chronic idiopathic disorder of the CNS sustained by a multifocal inflammatory process involving both the white and gray matters [16–18]. Although there are several forms of MS, during the initial phases of the disease a large proportion of patients present a relapsing–remitting (RR) clinical form of the disease (RR-MS). Currently, various disease-modifying drugs are indicated as first-line therapy for RR-MS. These include glatiramer acetate, interferon (IFN)-β1b and IFN-β1a, which are all widely used as first-line disease-modifying drugs [19–23]. However, their subcutaneous injection is hindered by possible local and systemic adverse effects (injection site pain, burning, myalgia, erythema, and necrosis) that affect patient compliance and thus efficacy [24–26].
The analgesic and anti-inflammatory properties of PEA make it an ideal candidate as adjuvant therapy to reduce the side effects of the immunomodulatory drugs available for RR-MS. Moreover, PEA was shown to reduce immune cells activation and proinflammatory cytokine expression in the CNS of mice with virus-induced MS [27]. Therefore, PEA could act by cooperating with immunomodulatory drugs and increase their efficacy in reducing pain and inflammation in MS.
Thus, the aim of the present study was to evaluate the oral administration of ultramicronized PEA (um-PEA) as an add-on therapy for RR-MS in reducing the IFN-β1a-related side effects, as well as the safety and tolerability of this adjuvant treatment.
Patients and Methods
Study Population
Twenty-nine patients (17 men, 12 women; mean ± SD age 27 ± 8 years) affected by definite RR-MS according to McDonald’s 2010 criteria [28], who were treated with subcutaneous IFN-β1a for at least 6 months and were experiencing IFN-β1a-related AEs, were enrolled at the Department of Neurosciences, Reproductive and Odontostomatological Sciences, Federico II University of Naples, Italy, from January 2011 to April 2011. Inclusion criteria were: definite diagnosis of RR-MS, age between 18 and 55 years, disease duration of < 1 year, and an Expanded Disability Status Scale (EDSS) score between 1.0 and 3.5. The exclusion criteria were: MS-related conditions, such as current clinical relapse (defined as newly developing symptoms or reactivation of pre-existing deficits for a minimum of 24 h occurring at least 30 days after the preceding episode, in the absence of increased body temperature or infections) [28], steroid therapy 30 days prior to study entry, concomitant diseases precluding the IFN-β1a treatment; pregnancy or breastfeeding, cognitive decline preventing informed consent, pathological conditions interfering with MS evolution, nonsteroidal anti-inflammatory drugs allergy, or intolerance to IFN-β1a. This study was approved by the institutional ethics committee for biomedical activities "Carlo Romano" Medical School Federico II University, Naples, Italy (registration number 197/2011), and all patients gave their written informed consent. The study was performed in compliance with the good clinical practice guidelines and the principles of the Declaration of Helsinki.
Experimental Design
This was a double-blind, randomized, placebo-controlled study. After the screening period, eligible patients were randomized 1:1 to receive ultramicronized PEA (um-PEA) (NORMAST® 600 mg/day/p.o.) or placebo, both added to subcutaneous IFN-β1a (Rebif® 44 μg; 3 times a week) administered using a prefilled syringe, for 12 months. Randomization was performed at our study center using kit numbers as randomization numbers. The study medication, um-PEA and placebo, were provided by Epitech Group SpA (Saccolongo, Italy) in the form of um-PEA or placebo-containing pills. All the pill containers were sequentially numbered, tamper-proof, equal in weight, and similar in appearance. All the patients enrolled in the study were blind to the treatment (PEA or placebo). All patients underwent a full neurological examination by a single neurologist during the screening phase and after 1, 6, and 12 months of treatment. This examination included: 1) the evaluation of motor disability using the EDSS [29]; 2) evaluation of quality of life (QoL) by the Multiple Sclerosis Quality of Life-54 questionnaire (MSQoL-54) (developed by combining the most widely utilized generic measure of quality of life in the world, the Short Form-36, with additional items specific to MS) [30]; 3) paced auditory serial addition test (PASAT) (a measure of cognitive function that assesses auditory information processing speed and flexibility, as well as calculation ability) [31, 32]; and 4) the assessment of pain using a visual analog scale (VAS) [generally presented as a single line of 100 mm with anchor words at either end (e.g., no pain to worst possible pain)] [33]. The width (in mm) of the erythema induced by IFN-β1a at its injection site was evaluated over its maximum diameter at 1 month, and after 6 and 12 months of PEA or placebo treatment. Blood samples were collected from all the patients to quantify NAE plasma levels (1, 3, 6, 9, and 12 months) and proinflammatory cytokines (1, 3, 6, and 12 months). The primary endpoints of the study were to evaluate the efficacy of um-PEA in 1) reducing IFN-β1a-related adverse effects in patients with RR-MS (pain and erythema width at the injection site); and 2) improving the patients’ QoL. The secondary endpoints were to evaluate the effects of um-PEA on 1) the serum profile of the proinflammatory cytokines IFN-γ, tumor necrosis factor-α (TNF-α), and interleukin (IL)-17; 2) the plasma levels of PEA, oleoylethanolamide (OEA), and the endocannabinoid anandamide (AEA); 3) clinical disease progression (sustained disability progression is clinically defined as at least a 1-point increase from baseline EDSS score); and 4) on safety and tolerability of um-PEA.
Quantification of the NAE plasma levels
Blood (4 ml) was collected in ethylenediaminetetraacetic acid-containing tubes (Becton Dickinson, Oxford, UK) on ice, and centrifuged (10 min; 4 °C; 2000 × g) within 1 h of collection. Aliquots (0.5 ml) of plasma were transferred into 1.5-ml tubes (Eppendorf, Hamburg, Germany) and immediately stored at –80 °C until assay. Only samples without evidence of hemolysis were used. For NAE extraction from plasma, we transferred 250 μl of plasma into a Sovirel tube containing the internal standards [d4-PEA (50 ng/ml), d4-OEA (50 ng/mL), and d4-AEA (10 ng/mL); all Cayman Europe, Tallinn, Estonia]. 2 ml of ethyl acetate-hexane (9:1, v/v) was then added and the mixture extracted for 30 min before centrifugation (2000 × g, 15 min). The organic layer was recovered and evaporated, in part, under N2 at 40 °C. The residual organic phase was transferred to a microvial and subsequently evaporated under N2 at 40 °C. The residue was finally reconstituted in 200 μl acetonitrile. The samples (8 μl) were analyzed by ultra performance liquid chromatography (UPLC)/tandem mass spectrometry using an Acquity UPLC system (Waters, Etten-Leur, the Netherlands) coupled to a TQD mass spectrometer (Waters) controlled by MassLinkx. Analyte separation was performed on a reverse-phase C18 column (Restek 100 mm × 2.1 mm i.d., 1.9 μm particle size) and a precolumn (Restek Ultra C18 10 mm × 2.1 mm i.d.) maintained at 40 °C with a mobile phase flow rate of 0.2 ml/min. Mobile phase A consisted of 10 mM ammonium formate in UPLC-grade water and 0.1 % (v/v) formic acid. Mobile phase B was UPLC-grade acetonitrile (Sigma-Aldrich, Milan, Italy) containing 0.1 % (v/v) formic acid. The total run time was 5 min. Electrospray ionization was carried out in the positive mode using nitrogen as the nebulizing gas. MS parameters for all analyses were established by infusing standards (100 ng/ml) using an infusion pump. The multiple reaction-monitoring modes were employed for quantification. Mass spectrometric parameters used for the analysis of AEA, PEA, and OEA are summarized in Supplementary Table 1 (Supplementary methods). The selected transitions for the quantitative measurements were as follows: AEA = 348.3 → 62.1; d4-AEA = 352.3 → 66.0; PEA = 300.3 → 62.0; d4-PEA = 304.3 → 62.0; OEA = 326.3 → 62.1; d4-OEA = 330.3 → 66.0. The calibration curves were constructed using linear regressions between the peak area ratios of each compound relative to the internal standard and the corresponding concentrations. Linearity for all analytes was confirmed in the examined ranges (R 2 ≥ 0.9985 for AEA; R 2 ≥ 0.9885 for PEA; R 2 ≥ 0.9958 for OEA). The validation parameters of the analytical method are reported in the Supplementary methods.
Cytokine Analysis
Serum levels of IFN-γ, TNF-α, and IL-17 were measured by enzyme-linked immunosorbent assays using predesigned kits from R&D Systems (DuoSet; R&D systems, Minneapolis, MN, USA), according to the manufacturer’s instructions. Briefly, microtiter plates (Nunc, U16; Maxisorp, Vedbaek, Denmark) were coated with monoclonal capture antibody (antihuman IFN-γ, TNF-α, or IL-17) and incubated at 4 °C overnight. The plates were then washed and subsequently incubated for 2 h with serum. The biotinylated detection antibody was then added and incubated for 2 h, followed by an incubation for 20 min at room temperature with streptavidin–horseradish peroxidase conjugate and the chromogenic substrate 3,3’,5,5’-tetramethylbenzidine (Bangalore Genie, Bangalore, India). The reaction was stopped using sulfuric acid (2 N) and the optical density reading was taken at 450 nm using an Teknika Microwell system, Reader 230 s (Organona, Eppelheim, Germany). All the experiments were conducted in duplicates. Standard curves were obtained using the standards provided by the manufacturer, and the results are expressed as concentration of cytokines (pg/ml).
Collection of Peripheral Blood Mononuclear Cells
Peripheral blood mononuclear cells (PBMCs) were isolated from patients with RR-MS at the study onset and every 3 months after therapy started. Peripheral blood was collected into ethylenediaminetetraacetic acid-anticoagulant vacutainer tubes. PBMCs were isolated by density-gradient centrifugation using Ficoll-Hypaque (GE Healthcare, Little Chalfont, UK). Mononuclear cells were harvested from the interface, washed 3 times with saline, and subsequently washed with red blood cell lysis buffer. Finally, the platelets were eliminated by an additional wash and centrifugation at 200 × g for 10 min. PBMCs were stored as pellets at −80 °C for mRNA isolation.
Fatty Acid Amide Hydrolase and N-acylethanolamine- hydrolyzing Acid Amidase mRNA Expression by PBMCs
mRNA extraction and quantitative reverse transcription polymerase chain reaction were performed as previously described [34]. Briefly, total RNA was extracted using the TriPure reagent (Roche, Basel, Switzerland), according to the manufacturer’s instructions. cDNA was synthesized using a reverse transcription kit (Promega, Leiden, the Netherlands) from 1 μg total RNA. Quantitative PCR was performed with a STEP one PLUS instrument and software (Applied Biosystems, Foster City, CA, USA). Data were normalized to the 60S ribosomal protein L19 (RPL19) mRNA expression, using the ∆∆Ct method. Primer sequences were as follows: RPL19 forward CACATCCACAAGCTGAAGGCA, reverse CTTGCGTGCTTCCTTGGTCT; fatty acid amide hydrolase (FAAH) forward CACACGCTGGTTCCCTTCTT, reverse GGGTCCACGAAATCACCTTTGA; N-acylethanolamine-hydrolyzing acid amidase (NAAA) forward ATGGAGCGTGGTTCCGAGTT, reverse AGGCTGAGGTTTGCTTGTCCT.
Safety Assessment
Safety and tolerability were assessed by evaluating AEs and clinical laboratory data (e.g., serum biochemistry and liver enzymes). Blood samples for the evaluation of clinical laboratory data were collected from all patients at each month of the treatment.
Statistical Analysis
Data were analyzed with Generalized Linear Mixed Model (GLMM) performed using SAS software version 9.2 (SAS Institute, Cary, NC, USA). Statistical results were considered statistically significant with p-values < 0.05. All variables analyses were conducted using the unpaired t test, Mann–Whitney test, χ2, or Fisher’s exact test when appropriate. Statistical tests were 2-tailed, and Bonferroni correction was applied to identify significant differences among groups. Sample size was not calculated as the trial was intended as a pilot study. The Spearman correlation coefficient was used for the correlation between PEA plasma levels, NAE, and cytokine serum levels.
Results
PEA Reduces Pain Perception Without a Reduction of the Erythema at IFN-β1a Injection Site
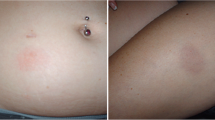
Demographics and baseline status showed no significant differences between the groups (Table 1). Upon treatment, the um-PEA-treated group perceived a significant improvement in pain sensation at the IFN-β1a injection site after 6 and 12 months compared with the control group (Fig. 1A). However, the improvement of pain sensation was not associated with a reduction in erythema width at the injection site compared with study onset, and no significant differences were observed between PEA-treated and placebo groups (Fig. 1B).

Ultramicronized palmitoylethanolamide (PEA) treatment reduces pain perception but not erythema at interferon (IFN)-β1a injection site. (A) Pain perception at the injection site of IFN-β1a was assessed using the visual analog scale at month 1, and at 6 and 12 months after starting PEA treatment (600 mg/day, p.o.). A significant effect of the treatment was found both at 6 and 12 months. Data are expressed as mean ± SEM; *p < 0.05 versus the placebo group. (B) The diameter of the erythema consequent to IFN-β1a injections was measured at 1 month, and after 6 and 12 months of PEA treatment. PEA had no effect on the erythema width
PEA Improves Cognitive Domain of QoL Without Affecting Clinical Disease Progression
The MSQoL-54 questionnaire evidenced that the um-PEA-treated group experienced a significant improvement in cognitive function, and change in health during the study, as shown in Table 2. We did not find any significant difference in the PASAT test between the PEA and placebo groups (Table 3). The neurological examination showed no significant progression of the disease in both groups, as evidenced by the absence of significant changes in the EDSS scores over the duration of the study (Table 4). We did not observe any incidence of treatment-emergent AEs (i.e., AEs that occurred after the first dose of the study drug). Similarly, we did not observe any clinically significant abnormalities in laboratory parameters in either group (data not shown). This shows the good tolerability of um-PEA treatment in patients with MS.
PEA Treatment Decreases Proinflammatory Cytokine Serum Levels
Although all the patients were receiving IFN-β1a, we decided to investigate the effects of um-PEA administration on the inflammatory markers. Indeed, PEA is a known anti-inflammatory bioactive lipid and its administration could have an impact on inflammation. Therefore, we quantified the serum levels of IFN-γ, IL-17, and TNF-α, 3 key cytokines known to play a role in the physiopathology of MS. We observed that um-PEA treatment induces a significant reduction in the serum levels of these proinflammatory cytokines (Fig. 2). Three months after the initiation of um-PEA treatment, the levels of IFN-γ and IL-17 were significantly reduced compared with placebo (effects of treatment). This decrease was maintained throughout the study (impact of time) (Fig. 2A, B). Similarly, TNF-α levels were significantly decreased by um-PEA after 6 and 12 months of treatment (Fig. 2C). The levels of IFN-γ, TNF-α, and IL-17 in the group receiving placebo remained relatively stable throughout the duration of the study.

Ultramicronized palmitoylethanolamide (PEA) treatment reduces proinflammatory cytokine serum levels. (A) Interferon (IFN)-γ, (B) interleukin (IL)-17, and (C) tumor necrosis factor (TNF)-α serum levels were measured by enzyme-linked immunosorbent assay at 1 month, and after 3, 6, and 12 months of treatment with either placebo or um-PEA (600 mg/day, p.o.). Similar cytokine levels between groups were found at 1 month, whereas at the subsequent time points PEA treatment reduced the cytokine levels compared with the placebo group. Data are expressed as mean ± SEM; ***p < 0.001, **p < 0.01, *p < 0.05 versus the placebo group
PEA Treatment Increases Circulating NAE Levels
As the treated patients received oral um-PEA, we asked whether this results in higher PEA plasma levels. As shown in Fig. 3A, um-PEA administration results in an increase of PEA plasma levels at month 3, which remained relatively stable over the treatment period and was significantly higher than the levels found in the placebo group. Because the hydrolytic pathways of PEA are shared by the other NAEs [2, 35], we next sought to determine if treatment with oral um-PEA also affected the levels of OEA and of the endocannabinoid AEA. As illustrated in Fig. 3(B, C), um-PEA treatment induced a significant increase in AEA and OEA plasma levels after 3 months, which persisted throughout the duration of the study. Of note, in the placebo group NAE plasma levels were not significantly altered throughout the study. Interestingly, when comparing the NAE plasma levels, we found a strong positive correlation between PEA and AEA plasma levels at 3, 6, 9, and 12 months of treatment (Table 5). OEA levels were also correlated with PEA levels, although less markedly so.

Ultramicronized palmitoylethanolamide (PEA) treatment increases plasma N-acylethanolamine levels. (A) PEA, (B) anandamide (AEA), and (C) oleoylethanolamide (OEA) plasma levels were measured by ultra performance liquid chromatography–mass spectrometry at 1 month, and after 3, 6, and 12 months of treatment with either placebo or um-PEA (600 mg/day, p.o.). At treatment onset, similar levels were found between the 2 treatment groups. After 3 months of treatment PEA, AEA, and OEA levels were increased and remained higher for the duration of the study. Data are expressed as mean ± SEM; ***p < 0.001, **p < 0.01 versus the placebo group
FAAH and NAAA mRNA Expression in PBMCs
The inactivation of NAEs (AEA, OEA, and PEA) occurs essentially by enzymatic hydrolysis by FAAH and NAAA [35–39]. Thus, it is possible that um-PEA administration reduces the degradation of AEA and OEA by substrate competition. It is also conceivable that the increased levels of PEA upon um-PEA administration have an impact on the expression of these two hydrolytic enzymes (Fig. 3A). Thus, our next step was to investigate FAAH and NAAA mRNA expression in PBMCs. We conducted this analysis in PBMCs because these cells are readily accessible and have been shown to mirror CNS changes of NAEs levels [40]. At 1 month of treatment, FAAH and NAAA expression was similar between the groups, as evidenced by a similar ΔCT (relative to the housekeeping gene RPL19) for the placebo and PEA groups for each enzyme (Fig. 4A). However, we observed a higher expression of NAAA than FAAH in each group, as evidenced by the lower ΔCT of NAAA (Fig. 4A). This higher expression of NAAA in PBMCs is reminiscent of its strong expression in immune cells. At end of the study, the data showed a moderate but significant difference in the expression of FAAH between the groups. Indeed, there was a slight increase in FAAH expression over time in the placebo group that was not present in the PEA-treated group (Fig. 4B). As for NAAA expression there was no significant change in expression over time or due to the treatment (Fig. 4C).

mRNA expression of the hydrolytic enzymes of N-acylethanolamines, fatty acid amide hydrolase (FAAH) and N-acylethanolamine- hydrolyzing acid amidase (NAAA) in peripheral blood mononuclear cells (PBMCs). FAAH and NAAA mRNA expression in PBMCs was measured by quantitative reverse transcription using RPL19 as housekeeping gene. (A) Expression of FAAH and NAAA at 1 month of treatment showing a higher expression of NAAA than FAAH, as evidenced by the smaller ΔCt numbers (the smaller the ΔCt number the higher the expression). (B) FAAH and (C) NAAA mRNA expression during the study. Data are expressed as mean ± SEM versus the placebo group. *p < 0.05 versus the placebo group
NAE Plasma Levels are Inversely Correlated with Proinflammatory Cytokines
Finally, because proinflammatory cytokine levels were decreased while NAE levels were increased by um-PEA treatment, we asked whether there was a correlation between the cytokine and bioactive lipid levels. We found, within 3 months of treatment with um-PEA, an inverse correlation between PEA and IFN-γ levels (Table 5). However, we did not find any correlation between the levels of PEA and those of TNF-α or IL-17. Interestingly, however, AEA plasma levels were inversely correlated with IFN-γ, TNF-α, and IL-17 serum levels [AEA vs IFN-γ: r = –0.7103 (p = 0.0004); AEA vs TNF-α: r = –0.6042 (p = 0.0048); AEA vs IL-17: r = –0.5506 (p = 0.0012)]. We also performed a hierarchical clustering analysis to investigate whether we could distinguish the um-PEA-treated patients from the placebo-treated patients only based on their cytokine (IFN-γ, IL-17, TNF-α) serum levels. While at baseline no particular cluster was apparent, a progressive clusterization appears over time (starting from month 3 to month 12). Actually, after 12 months of treatment, we found a nearly perfect clusterization between the placebo and the um-PEA-treated group (Fig. 5), thus further supporting the beneficial effects of um-PEA administration on the inflammatory tone of these patients.

Cluster analysis of patients after 12 months of treatment. Hierarchical cluster analysis of patients based on their circulating tumor necrosis factor (TNF)-α, interleukin (IL)-17, and interferon (IFN)-γ levels measured at 12 months of treatment. The patients that received placebo are represented in blue and those treated with um-PEA are represented in red
Discussion
The results of the present study describe, for the first time, the effects of oral um-PEA treatment as add-on therapy to IFN-β1a in a cohort of patients with RR-MS.
Pain is an important component of MS, which, despite its significant impact on patients’ QoL, is often neglected or undertreated. Patients with MS present not only spontaneous pain, but also several forms of evoked pain such as cutaneous mechanical pain and cold hypersensitivity at the distal extremities [41, 42]. Promising results in the treatment of pain have been reported for PEA in various clinical trials. In a pivotal randomized, double-blind, placebo-controlled clinical trial of 636 patients with sciatic pain, a significant and clinically relevant analgesic effect was documented for 300 mg and 600 mg PEA [43]. Hesselink and Hekker [44] also reported a series of clinical cases describing the potential efficacy and safety of um-PEA in the treatment of various syndromes associated with chronic pain. In the present study, patients treated with 600 mg um-PEA perceived an improvement in pain sensation at the injection site of IFN-β1a. In addition to pain, a common adverse effect of IFN-β1a therapy is the erythema developing at the injection site. This reaction is currently thought to represent a local inflammatory response to the injected drug, which is influenced by the injection path and depth. This is consistent with the observation that skin reaction is more likely to occur after injection in the arms or thighs, and less frequently in areas with a higher proportion of subcutaneous fat (e.g., abdomen or buttocks). Our analysis showed that, despite the reduced circulating cytokine levels, the treatment with um-PEA was not associated with a decrease of the erythema at the injection site (abdomen and arms). However, this might be owing to the fact that erythema is a highly local reaction to injection that might better respond to local administration of um-PEA.
QoL is a crucial element for patients living with MS. Although the PASAT test did not show any improvement, we found a positive effect of PEA treatment in the cognitive function and health in the MSQoL questionnaire. This result is in line with recent preclinical studies, showing that NAEs may exert a beneficial effect following cognitive impairments [45–47].
Unfortunately, the relatively small sample size of this study did not allow us to assess the relationship between clinical disease activity measures and magnetic resonance imaging. Nevertheless, we showed that PEA treatment reduces IL-17 levels in patients with RR-MS treated with IFN-β1a. This is significant as IL-17 mediates the inflammatory response in MS by controlling the recruitment of immune cells and by regulating the production of proinflammatory cytokines [48, 49]. Indeed, IL-17 induces the expression of TNF-α and chemokines, attracts neutrophils, and enhances the maturation of dendritic cells [50, 51]. Consistent with the reduced IL-17 serum levels in the um-PEA-treated group, we also found reduced TNF-α serum levels. Importantly, this effect is obtained under IFN-β1a treatment, which per se reduces the proinflammatory cytokine serum levels in MS [52–54]. Inflammatory mediators, such as IFN-γ and TNF-α are known key contributors to the induction and maintenance of autoimmune diseases [55–58]. However, although there is a vast literature regarding the anti-inflammatory effects of PEA in murine models, our study is the first to show the ability of this bioactive lipid to reduce the proinflammatory cytokine profile in RR-MS.
We also found in this study that PEA levels are significantly correlated with AEA and OEA levels. It remains to be determined whether this is due to competition for their hydrolysis or to changes in the expression of the hydrolytic enzymes, or both. Here we did not find any strong effect of um-PEA treatment on NAAA mRNA expression in PBMCs. However, after a year of treatment, PEA prevented the IFN-β1a-induced increase in FAAH mRNA expression (Fig. 4B). This is important as it was shown in human PBMCs that FAAH mRNA expression correlates with protein expression and activity [59, 60]. In fact, here we found that IFN-β1a treatment results in decreased AEA levels after 12 months of treatment [“IFN-β1a + placebo group”; 2-way analysis of variance followed by the Bonferroni post-test (p < 0.01 for 12 months vs 1 month)]. However, the fact that AEA and OEA levels are already increased after 3 months of um-PEA treatment might point towards a competition of AEA and OEA with PEA for their hydrolysis, as previously demonstrated in other settings.
The fact that oral administration of um-PEA not only results in increased plasma levels of PEA, but also of other NAEs is interesting in terms of potential synergistic effects, as the NAEs do not all share the same molecular targets. Indeed, most of the effects of PEA on inflammation and pain are mediated by PPAR-α [5, 11, 13, 61], whereas AEA mainly acts by activating cannabinoid receptors 1 and 2 [62, 63], and vanilloid (transient receptor potential vanilloid 1) receptors [64–66], and OEA by activating G protein-coupled receptor 119 and PPAR-α [67, 68]. Thus, the effects observed here upon um-PEA administration can be due to PEA activating its molecular targets, and also to PEA favoring the actions of other NAEs (e.g., AEA and OEA). Indeed, anti-inflammatory, neuroprotective, and analgesic effects have been reported for AEA and OEA in several preclinical models [69–73].
In conclusion, although the sample size is relatively small and the exact mechanisms explaining the effects of um-PEA are yet to be determined, we show here that oral administration of um-PEA as an add-on therapy in patients with RR-MS provides beneficial effects in terms of reduced circulating proinflammatory cytokines, pain sensation, and increased NAE levels.
References
Cadas H, Schinelli S, Piomelli D. Membrane localization of N-acylphosphatidylethonolamine in central neurons: studies with exogenous phospholipases. J. Lipid Mediat Cell Signal 1996;14:63–70.
Alhouayek M, Muccioli GG. Harnessing the anti-inflammatory potential of palmitoylethanolamide Drug Discov Today 2014;19:1632-1639.
Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature 1998;394:277-281.
Calignano A, La Rana G, Piomelli D. Antinociceptive activity of the endogenous fatty acid amide, palmitoylethanolamide. Eur J Pharmacol 2001;419:191-198.
Lo Verme J, Russo R, La Rana G, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther 2006;319:1051-1061.
Lambert DM, Vandevoorde S, Diependaele G, Govaerts SJ, Robert AR. Anticonvulsant activity of N-palmitoylethanolamine, a putative endocannabinoid, in mice. Epilepsia 2001;42:321-327.
Esposito E, Impellizzeri D, Mazzon E, Paterniti I, Cuzzocrea S. Neuroprotective activities of palmitoylethanolamide in an animal model of parkinson’s disease. Plos One 2012;7:e41880.
D'Agostino G, Russo R, Avagliano C, Cristiano C, Meli R, Calignano A. Palmitoylethanolamide protects against the amyloid-β25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 2012;37:1784-1792.
Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon A. N-(2-Hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur J Pharmacol 1996;300:227-236.
Esposito E, Paterniti I, Mazzon E, et al. Effects of palmitoylethanolamide on release of mast cell peptidases and neurotrophic factors after spinal cord injury. Brain Behav Immun 2011;25:1099-1112
Lo Verme J, Fu J, Astarita G, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol 2005;67:15-19
D'Agostino G, La Rana G, Russo R, et al. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther 2007;322:1137-1143
D'Agostino G, La Rana G, Russo R, et al. Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF-kappaB nuclear signaling in dorsal root ganglia. Eur J Pharmacol 2009;613:54-59
Lo Verme J, La Rana G, Russo R, Calignano A, Piomelli D. The search for the palmitoylethanolamide receptor. Life Sci 2005;77:1685-1698
Solorzano C, Zhu C, Battista N, et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci USA 2009;106:20966-20971.
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG Multiple sclerosis. N Engl J Med 2000;343:938-952
Chard D, Miller D. Grey matter pathology in clinically early multiple sclerosis: evidence from magnetic resonance imaging. J Neurol Sci 2009;282:5-11
Calabrese M, Filippi M, Rovaris M, et al. Morphology and evolution of cortical lesions in multiple sclerosis. A longitudinal MRI study. Neuroimage. 2008;42:1324-1328
Bornstein M, Miller A, Slagle S, et al A pilot trial of COP 1 in exacerbating-remitting multiple sclerosis. N Engl J Med. 1987;317:408-414.
The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing remitting multiple sclerosis. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43:655-661
Jacobs L, Cookfair D, Rudick R. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996;39:285-294.
PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352:1498-1504.
Johnson KP, Baringer JR. Current therapy of multiple sclerosis. Hosp. Pract. 1995;36:21-2, 25-8
Walther EU, Hohlfeld R. Multiple sclerosis: side effects of interferon beta therapy and their management. Neurology. 1999;53:1622-1627.
Panitch H, Goodin DS, Francis G, et al. EVIDENCE Study Group. Evidence of Interferon Dose-response: Europian North American Compartative Efficacy; University of British Columbia MS/MRI Research Group. Randomized, comparative study of interferon beta-1a treatment regimens in MS: The EVIDENCE Trial. Neurology. 2002;59:1496-1506.
Carotenuto A, Iodice R, Barbato F, Orefice NS, Orefice G. Necrotizing skin lesion and radial nerve palsy in a patient treated with glatiramer acetate. J. Neurol. Sci. 2013;331:172-173.
Loria F, Petrosino S, Mestre L, et al. Study of the regulation of the endocannabinoid system in a virus model of multiple sclerosis reveals a therapeutic effect of palmitoylethanolamide. Eur. J. Neurosci. 2008;287:633-641
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011;69:292-302.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis. An expanded disability status scale (EDSS). Neurology, 1983;33:1444-1452
Vickrey BG, Hays RD, Harooni R, Myers LW, Ellison GW. A health-related quality of life measure for multiple sclerosis. Qual. Life Res. 1995;4:187-206.
Forn C, Belenguer A, Parcet-Ibars MA, Avila C. Information-processing speed is the primary deficit underlying the poor performance of multiple sclerosis patients in the Paced Auditory Serial Addition Test (PASAT). J. Clin. Exp. Neuropsychol. 2008;30:789-796.
Fos LA, Greve KW, South MB, Mathias C, Benefield H. Paced visual serial addition test: an alternative measure of information processing speed. Neuropsychology. 2000;7:140-146.
Grau-López L, Sierra S, Martínez-Cáceres E, Ramo-Tello C Analysis of the pain in multiple sclerosis patients. Neurologia. 2011;26:208-213.
Alhouayek M, Masquelier J, Cani PD, Lambert DM, Muccioli GG Implication of the anti-inflammatory bioactive lipid prostaglandin D2-glycerol ester in the control of macrophage activation and inflammation by ABHD6. Proc. Natl. Acad. Sci. USA. 2013;110:17558-17563
Muccioli GG. Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discov. Today. 2010;15:474-483
Cravatt BF, Demarest K, Patricelli MP, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9371-976.
Ahn K, Johnson DS, Mileni M, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009;16:411-420.
Sun YX, Tsuboi K, Zhao LY, Okamoto Y, Lambert DM, Ueda N. Involvement of N-acylethanolamine-hydrolyzing acid amidase in the degradation of anandamide and other N-acylethanolamines in macrophages. Biochim. Biophys. Acta 2005;1736:211-220.
Ueda N, Tsuboi K, Uyama T. Enzymological studies on the biosynthesis of N-acylethanolamines. Biochim. Biophys. Acta 2010;1801:1274-1285.
Centonze D, Battistini L, Maccarrone M. The endocannabinoid system in peripheral lymphocytes as a mirror of neuroinflammatory diseases. Curr. Pharm. Des. 2008;14:2342-2370.
O’Connor AB, Schwid SR, Herrmann DN, Markman JD, Dworkin RH. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain 2008; 137:96-111
Foley PL, Vesterinen HM, Laird BJ, et al. Prevalence and natural history of pain in adults with multiple sclerosis: systematic review and meta-analysis. Pain. 2013;154(5):632-642
Guida G, de Martino M, de Fabiani A, et al. La palmitoiletanolamida (Normast) en el dolor neuropatico cronico por lumbociatalgia de tipo compresivo: estudio clinico multicentrico. [Palmitoylethanolamide treatment of sciatic pain: results form a multicenter study]. Dolor. 2010; 25:35-42.
Hesselink JM, Hekker TA Therapeutic utility of palmitoylethanolamide in the treatment of neuropathic pain associated with various pathological conditions: a case series. J Pain Res. 2012;5: 437-442.
Hao S, Avraham Y, Mechoulam R, et al. Low dose anandamide affects food intake, cognitive function, neurotransmitter and corticosterone levels in diet-restricted mice. Eur J Pharmacol. 2000;392(3):147-156.
Barbierato M, Facci L, Marinelli C, et al. Co-ultramicronized palmitoylethanolamide/luteolin promotes the maturation of oligodendrocyte precursor cells. Sci Rep. 2015;5:16676.
Yang LC, Guo H, Zhou H, et al. Chronic oleoylethanolamide treatment improves spatial cognitive deficits through enhancing hippocampal neurogenesis after transient focal cerebral ischemia. Biochem Pharmacol. 2015;94:270-281.
Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232-239.
Zhu S, Pan W, Song X, et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat. Med. 2012;18:1077-1086.
Meares GP, Ma X, Qin H, Benveniste EN. Regulation of CCL20 expression in astrocyte by IL-6 and IL-17 Glia 2012;60:771-781
Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467-476.
Chen M, Chen G, Nie H, et al. Regulatory effects of IFN-beta on production of osteopontin and IL-17 by CD4+ T cells in MS. Eur. J. Immunol. 2009;39:2525-2536.
Christophi GP, Panos M, Hudson CA, et al. Interferon-beta treatment in multiple sclerosis attenuates inflammatory gene expression through inducible activity of the phosphatase SHP-1. Clin Immunol. 2009; 133:27-44.
Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasone activation. Immunity. 2011;34:213-223
Lucchinetti CF, Parisi J, Bruck W. The pathology of multiple sclerosis. Neurol Clin 2005;23:77-105.
Lassmann H. Multiple sclerosis pathology: evolution of pathogenetic concepts. Brain Pathol. 2005;15:217-222.
Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol 2007;17:210-218.
Lassmann H. Mechanisms of inflammation induced tissue injury in multiple sclerosis. J. Neurol. Sci. 2008;274:45-47.
D'Addario C, Di Francesco A, Arosio B, et al. Epigenetic regulation of fatty acid amide hydrolase in Alzheimer disease. PLoS One 2012;7:e39186.
Wolfson ML, Aisemberg J, Salazar AI, et al. Progesterone reverts LPS-reduced FAAH activity in murine peripheral blood mononuclear cells by a receptor-mediated fashion. Mol Cell Endocrinol. 2013;381:97-105.
Scuderi C, Steardo L. Neuroglial roots of neurodegenerative disease: therapeutic potential of palmitoylethanolamide in models of Alzheimer’s disease. CNS Neurol Disorder Drug Target 2013;12:62-69.
Eljaschewitsch E, Witting A, Mawrin C, et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. 2006;49:67-79.
Correa F, Docagne F, Mestre L, et al. A role for CB2 receptors in anandamide signalling pathways involved in the regulation of IL-12 and IL-23 in microglial cells. Biochem Pharmacol. 2009;77:86–100.
De Petrocellis L, Davis JB, Di Marzo V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett 2001;506:253-256.
Di Marzo V, Blumberg PM, Szallasi A. Endovanilloid signaling in pain. Curr. Opin. Neurobiol. 2002;12:372–379.
Ross RA. Anandamide and vanilloid TRPV1 receptors. Br. J. Pharmacol. 2003;140:790-801
Fu J, Gaetani S, Oveisi F, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 2003;425:90-93.
Overton HA, Babbs AJ, Doel SM, et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006;3:167-175.
Baker D, Pryce G, Croxford JL, et al. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001;15:300-302.
Gubellini P, Picconi B, Bari M et al. Experimental Parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J. Neurosci. 2002;22: 6900-6907.
Bisogno T, Martire A, Petrosino S, Popoli P, Di Marzo V. Symptom-related changes of endocannabinoid and palmitoylethanolamide levels in brain areas of R6/2 mice, a transgenic model of Huntington’s disease. Neurochem Int. 2008;52:307-313.
Zhou Y, Yang L, Ma A, et al. Orally administered oleoylethanolamide protects mice from focal cerebral ischemic injury by activating peroxisome proliferator-activated receptor α. Neuropharmacol. 2012;63:242-249.
Gonzalez-Aparicio R, Blanco E, Serrano A, et al. The systemic administration of oleoylethanolamide exerts neuroprotection of the nigrostriatal system in experimental Parkinsonism. Int J Neuropsychopharmacol. 2014;17:455-468.
Acknowledgments
M.A. is a postdoctoral researcher from the FRS-FNRS (Fonds de la Recherche Scientifique) Belgium. G.G.M. is the recipient of grants from the Fondation Charcot and the FRS-FNRS, Belgium (grant 3.4521.10). We thank Dr. Carlo Schievano (Adjunct Professor of Padua University) for helping with the statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article. (PDF 2210 kb)
ESM 2
(DOCX 24 kb)
Rights and permissions
About this article

Cite this article
Orefice, N.S., Alhouayek, M., Carotenuto, A. et al. Oral Palmitoylethanolamide Treatment Is Associated with Reduced Cutaneous Adverse Effects of Interferon-β1a and Circulating Proinflammatory Cytokines in Relapsing–Remitting Multiple Sclerosis. Neurotherapeutics 13, 428–438 (2016). https://doi.org/10.1007/s13311-016-0420-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0420-z