
Abstract
Tuberous sclerosis complex (TSC) is a genetic disorder with a high prevalence of autism spectrum disorder (ASD). Tremendous progress in understanding the pathogenesis of TSC has been made in recent years, along with initial trials of medical treatment aimed specifically at the underlying mechanism of the disorder. At the cellular level, loss of TSC1 or TSC2 results in upregulation of the mechanistic target of rapamycin (mTOR) pathway. At the circuitry level, TSC and mTOR play crucial roles in axonal, dendritic, and synaptic development and function. In this review, we discuss the molecular mechanism underlying TSC, and how this disease results in aberrant neural connectivity at multiple levels in the central nervous system, leading to ASD symptoms. We then review recent advances in mechanism-based treatments of TSC, and the promise that these treatments provide for future mechanism-based treatment of ASD. Because of these recent advances, TSC represents an ideal model for how to make progress in understanding and treating the mechanisms that underlie ASD in general.
Similar content being viewed by others

Introduction and Background
Prevalence of Tuberous Sclerosis Complex and Autism Spectrum Disorder
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder due to mutations in either TSC1 or TSC2. Spontaneous genetic mutations occur in 2/3 cases. The incidence of TSC is about 1 in 6000 live births [1], and it presents with a wide range of manifestations caused by localized cellular overgrowth leading to benign tumors (hamartomas) in multiple organs, including the brain [cortical and subcortical tubers, subependymal nodules, and subependymal giant cell astrocytomas (SEGAs)], eye (retinal hamartomas), heart (rhabdomyomas), lungs (lymphangioleiomyomatosis), kidneys [angiomyolipomas], and skin (hypomelanotic macules, angiofibromas, and shagreen patches) [2–4]. Approximately 90 % of patients with TSC will have some level of tuberous sclerosis-associated neuropsychiatric disorder (TAND), including autism spectrum disorder (ASD), attention deficit hyperactivity disorder, depression and anxiety, intellectual disability, and specific learning disorders [5]. The prevalence of ASD in TSC varies depending on the sampled population, diagnosis definition, and testing methodologies used but ranges from 26 % to 50 % [6, 7]. TSC is one of the most frequently identified monogenic causes of ASD.
Advantages of TSC as a Disease Model to Study ASD
TSC provides a number of advantages as a model disorder in which to study early development and treatment of ASD. These include the possibility of pre- or neonatal identification of patients, the high prevalence of ASD among the TSC population, the phenotypic variability of TSC presentation, the current level of understanding and interest in the mechanisms underlying TSC, and the current progress in mechanism-based treatment for TSC [8]. Owing to the presence of cardiac rhabdomyomas being detected with ultrasound, many patients with TSC are now diagnosed prenatally or at birth [9]. Early diagnosis offers a chance to follow the development of ASD symptoms and biomarkers from the earliest possible time period, well before any neuropsychiatric symptoms become evident. The prevalence of ASD in patients with TSC is higher than in many other cohorts with the potential for presymptomatic identification, including siblings of children with ASD [6, 10]. The severity of ASD symptoms in patients with TSC varies from generally unaffected to severely affected, and ASD symptoms are accompanied by range of associated neuropsychological deficits. A recent study of ASD behavioral “signatures” found that out of a group of 6 genetic disorders associated with ASD, patients with TSC had the widest range of autistic features, and the most overlap with a sample of patients with idiopathic ASD [11]. This would indicate that findings related to ASD in TSC may have broad applicability to a range of patients with ASD of multiple causes. Some of the possible reasons underlying this variability in ASD symptoms and severity in TSC will be explored in the section “The Pathophysiology of ASD in TSC”.
One of the most compelling reasons to use TSC as a model in which to study ASD is that the cellular mechanisms at the root of the disorder have become increasingly well characterized over the last decade. The overactive mTOR pathway seen in TSC has been implicated in numerous other diseases, including cancer, obesity, type 2 diabetes, and neurodegenerative disorders, as well as other genetic disorders presenting with ASD, prompting intensive investigation into its normal function and pathological dysfunction [12–14]. Several mTOR-inhibiting drugs have been used and studied for some time in oncology and for immune suppression; more recently, these drugs have been approved for use in patients with TSC for the treatment of SEGAs and renal angiomyolipomas, and are being studied in the treatment of other manifestations of the disease, including neuropsychiatric symptoms [4, 7]. Understanding the cellular mechanisms of TSC and having a mechanism-based treatment opens a world of possibility for understanding and treating ASD “from the ground up”.
Mechanism of TSC: The mTOR Pathway
The mTOR pathway is a common intracellular biochemical pathway responsible for regulating mRNA translation, autophagy, stress pathways, and other functions related to cellular growth and homeostasis [13, 15, 16]. mTOR is a serine–threonine kinase that is an essential component of two complexes, mTOR complex 1 and 2 (mTORC1 and mTORC2). mTORC1 is regulated by the products of TSC1 and TSC2, hamartin and tuberin, respectively. These bind together along with TBC1D7 to form a heteromeric complex that acts as a GTPase-activating protein, which inactivates Ras homolog enriched in brain (Rheb). When Rheb is inactivated, the function of the mTORC1 complex is inhibited. The mTORC1 is a key regulatory complex that is controlled by extracellular signals that affect mRNA translation. In normally functioning cells, it is responsive to low energy levels (increased adenosine monophosphate/adenosine triphosphate ratio) via the adenosine monophosphate-dependent kinase, which then inhibits cell growth. mTORC1 is also activated by upstream activity of several growth factors on the TSC complex, including insulin and insulin-like growth factor 1, which stimulate the phosphoinositide 3-kinase and Ras pathways to trigger cell growth [12].
With loss of function of either TSC1 or TSC2 in TSC disease, mTORC1 becomes overactive, leading to phosphorylation of eIF4E-binding protein 1 and p70 S6 kinase 1, which activates mRNA translation. This leads to cellular overgrowth and metabolic overactivity, causing many of the multisystemic effects of TSC.
The Pathophysiology of ASD in TSC
The pathophysiology underlying ASD in patients with TSC is an area of intense interest and inquiry, and one in which attention to the mechanisms at play in TSC may shed some light. The core abnormality in TSC, dysregulation of the mTOR pathway, leads to abnormal brain development and functioning at all levels of neural function, from intracellular biochemistry to the brain as a whole. This includes abnormalities in intracellular signaling; cell growth and development; neuronal migration; and axon, dendrite, and synapse formation and maintenance. All of these mechanisms have been postulated to be part of the underlying pathophysiology leading to ASD. In TSC, these abnormalities lead to formation of dysplastic neurons, tubers, and aberrant neuronal connections, and presumably to the ASD, epilepsy, and other TAND symptoms seen in patients. This section will review recent findings related to TSC and autism due to these multilevel systemic defects.
Dysfunction of the mTOR Pathway Leading to Neuronal Intracellular Abnormalities
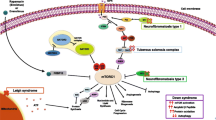
Loss of activity of either TSC1 or TSC2 leads to loss of inhibition of mTORC1 and increased downstream activity, including increased protein synthesis, without regard to upstream growth factors or energy availability (Fig. 1a). In neurons, this leads to dysregulation of a number of vital functions related to neuronal function and signaling, including axon specification and guidance, dendritic morphogenesis, and synapse formation and adaptation. Deficits in these fundamental processes can lead to abnormalities in neuronal circuit formation and activity-dependent plasticity. Additionally, dysfunction of the mTOR pathway leads to defects in autophagy and the cellular stress response [17–19], potentially increasing vulnerability to external stressors such as seizures, hypoxia, inflammation, and toxins. When the mTOR pathway is dysregulated in neuroglial progenitor cells, it results in abnormalities in brain development, including defects in cell growth, migration, lamination, and myelination [20]. Dysfunction of the mTOR pathway is also implicated in a number of other single-gene disorders with a high incidence of ASD, such as phosphatase and tensin homolog deleted in chromosome ten(PTEN)-associated disorders, neurofibromatosis type 1, and fragile X [14, 21, 22].

The multiple levels of neural structure and function affected in tuberous sclerosis complex (TSC). (a) Biochemical: dysregulation of the mechanistic target of rapamycin (mTOR) pathway leads to unchecked cellular growth. (b) Neuronal: cellular overgrowth causes abnormal synaptic excitability and long-term potentitation (LTP), dendritic spine morphology, neuronal structure, and cortical lamination. (c) Brain: cellular overgrowth and abnormal migration results in formation of tubers, abnormal white matter myelination and connectivity, and cerebellar abnormalities. (d) Organism: multiple neural abnormalities result in seizures, abnormal cognition, inflammation, and autism spectrum disorder symptoms. PI3K = phosphoinositide 3-kinse; AMP=adenosine monophosphate; ATP=adenosine triphosphate; AKT=protein kinase B; AMPK=AMP-activated protein kinase; mTORC1 = mTOR complex 1; 4E-BP1 = eIF4E-binding protein 1; S6K1 = p70 S6 kinase 1
Abnormalities of Synaptic Function and Intercellular Signaling
The cellular abnormalities resulting from mTOR dysregulation have a cascading effect that leads to defects in synapse formation and function (Fig. 1b). Recent studies have pointed to abnormalities at the level of the synapse in a number of conditions associated with ASD [23–25]. Upregulation of the mTOR pathway leads to abnormal dendritic protein synthesis with reduced or dysmorphic dendritic spines and alterations in postsynaptic glutamate receptor-mediated long-term depression [26–28]. These synaptic abnormalities have been postulated to contribute to deficits in learning, memory, and adaptation seen in ASD [15, 23, 29]. Changes at the synapse or electrical properties of the cells may then lead to abnormal neuronal excitability, which has been shown to be decreased in cerebellar Purkinje cells and increased in hippocampal pyramidal neurons of TSC mutant mice [28, 30]. In addition, decreased gamma-aminobutyric acid (GABA)-ergic inhibition and increased glutamatergic excitation is implicated in the increased seizure susceptibility seen in TSC and likely also contributes to cognitive dysfunction [31–33].
Abnormalities of Brain Development and Neural Connectivity
The mTOR pathway is active during embryonic brain development and affects the initial growth and development of neurons and glia. In addition to causing abnormalities and overgrowth of individual cells, mTOR dysregulation leads to abnormalities in neuronal migration and formation of cortical lamination (Fig. 1b). These migration abnormalities appear to be responsible for the various structural brain malformations characteristic of TSC, including tubers, white matter heterotopias, radial migration lines, and subependymal nodules (Fig. 1c) [15]. Studies of postmortem brains of patients with TSC have also shown microscopic structural abnormalities in grossly normal-appearing regions, potentially indicating more widespread abnormalities in brain structure than would be suggested by neuroimaging [34]. The variety of phenotypes seen in TSC may be a result of variations in location of tubers and other macrostructural abnormalities, with tubers particularly in temporal and cerebellar locations being correlated with ASD symptoms in TSC [35–37]. In addition to abnormalities in glial and neuronal migration, the formation of myelin also appears to be affected in TSC [34, 38], thus contributing to the white matter abnormalities found using diffusion-weighted imaging of patients with TSC. These patients had decreased anisotropy in regions of radiographically normal-appearing white matter, indicating abnormal white matter microstructure. This decrease was more severe in patients with TSC with an autism phenotype throughout the white matter and specifically in language-related pathways [39, 40]. The abnormal neuronal formation, migration, and wiring in TSC may result in the deficits in functional neural connectivity that have been postulated to be at the root of ASD [41–43]. Local overconnection may result from clusters of cells that do not properly migrate and laminate and form excessive axonal and dendritic connections. Long-range underconnection may result from disorders of myelination, axonal pathfinding defects and abnormal larger-scale neuronal migration.
Epilepsy, Development, and Environment Effects on ASD in TSC
A number of systemic factors and comorbid conditions affect the ASD phenotype and severity in TSC, including epilepsy, cognitive impairment, and inflammation (Fig. 1d). Epilepsy is the most prevalent neurologic comorbidity of TSC, present in about 85 % of patients, with the majority of patients developing seizures within the first year, and often within the first few months, of life. Infantile spasms are particularly common, although focal seizures also often occur before, after, or in conjunction with spasms [2, 3]. Studies have shown correlations with earlier onset of seizures and worse cognitive outcomes in rodents and humans [44, 45]. A study of early developmental trajectories in children with TSC and ASD found a trend towards more severe epilepsy in patients with TSC with ASD, as well as a decreased nonverbal intelligence quotient (IQ) in patients with longer seizure durations [10]. Early studies suggest treatment of epileptic abnormalities in TSC prior to the onset of seizures results in improved outcomes in intellectual ability and epilepsy control [46, 47]. This suggests that more severe or poorly controlled epilepsy may contribute to worse cognitive outcomes in TSC and ASD, and correlations to that effect have been shown, particularly with infantile spasms [48]. However, the correlative versus causal relationship between epilepsy and autism remains an area in need of further exploration [49, 50].
Patients with TSC have a high prevalence of cognitive impairment and intellectual disability, but there is a wide range of severity across patients. IQ scores in TSC appear to be roughly bimodally distributed, with one group of patients with severe intellectual disability and another group with intelligence in the normal range but with a downshifted mean [3]. The biological basis for this bimodal distribution remains unclear. Investigations of early developmental trajectories of children with TSC have shown correlations between cognitive impairment and autism, such that children with the most severe autism also have the most severe intellectual impairment. Even children with TSC but without an ASD diagnosis had impairment in their play behavior [6]. In addition, children with TSC and an ASD diagnosis had a decline in their nonverbal IQ, verbal IQ, and developmental quotient in the second to third year of life, even when controlling for the duration of seizures, while their non-ASD counterparts had gains in these domains [10]. The relationship between cognition and autism in TSC may be a result of a common underlying neurodevelopmental dysfunction, increased difficulty with learning due impaired social interactions, or some combination thereof.
The relationship between inflammation and autism is an active area of research, as well as one of controversy. One area of particular relevance to autism in TSC is the relationship between mTOR pathway dysregulation and altered immune system activity in patients with ASD [51]. Other studies have focused on the interaction between prenatal inflammatory triggers and TSC genetics, including the finding that mice with Tsc2 haploinsufficiency and intrauterine immune activation were more likely to show deficits in social behavior. This was paired with an observation that, when compared with children with TSC without ASD or children with idiopathic ASD, more children with both ASD and TSC had the third trimester of their development occur during a time of peak seasonal influenza activity [8]. These findings suggest that the interaction of increased immune activation during late intrauterine development with the TSC genotype may contribute to the development of ASD.
Promising Treatments
mTOR Inhibitors
The first mTOR inhibitor, rapamycin (sirolimus), was discovered in 1975 as an antifungal agent and was subsequently found to have antiproliferative and immunosuppressive properties, leading to its initial uses in preventing solid organ transplant rejection and restenosis of coronary arteries following angioplasty. Following the elucidation of the mTOR pathway and the role of TSC1 and TSC2, and with the development of derivative medications with improved pharmacokinetics and side effect profiles, over the last decade mTOR inhibitors have begun to be studied and used in treatment of the various manifestations of TSC, as well as in a number of malignancies (see [52] and [53] for reviews). Sirolimus and its derivatives (“rapalogs”) such as everolimus work by binding FKBP12 and then interacting with the FKBP12–rapamycin-binding domain of mTORC1, inhibiting the serine–threonine kinase activity of mTORC1 and preventing mRNA translation. This effectively restores the mTORC1 inhibition that would normally be provided by the TSC1/2 complex that is dysfunctional in patients with TSC. A second generation of mTOR kinase domain inhibiting medications with activity against both mTORC1 and mTORC2 are now under development and in early clinical trials [53]. With the clinical availability of these medications, studies of their use in TSC have shown a number of promising results in animal and human studies.
Therapeutic Effects of mTOR Inhibitor Treatment in Animal Models of TSC
A number of animal models of TSC have been developed, with both spontaneous and conditional knockout of either TSC1 or TSC2 in neural stem cells, neurons, or glial cells (Table 1). These animals develop most of the pathologic features of TSC, including abnormal neuronal migration and lamination, increased cell size, and hypomyelination, as well as a number of neurocognitive features, including seizures, impairment in learning and memory, and deficits in social behavior.
Trials using mTOR inhibitors in animal models of TSC have shown that blocking the action of mTOR can reverse a number of manifestations of TSC, both at the cellular level and in the behavior and survival of the animal. Zeng et al. [69] demonstrated in mice with conditional knockout of Tsc1 in glial cells that early treatment with rapamycin prevented the development of epilepsy, and later treatment decreased seizures and prolonged survival. In another mouse model with neuron-specific conditional knockout of Tsc1 in neuronal cells, treatment with rapamycin or everolimus normalized levels of mTOR pathway constituents with corresponding improvement in neurofilament abnormalities, reduction of enlarged cells, and restoration of myelination [66]. Treated animals showed significant improvement in seizures and overall survival, although these improvements were attenuated when treatment was discontinued. These improvements were seen despite no evidence in reversal of neuronal migration abnormalities and only minor improvement in dendritic spine density and length, indicating not all mTOR-related structural and functional abnormalities may be equally important for behavioral abnormalities or seizures, or that correction of some but not all abnormalities may be sufficient to improve outcome.
Ehninger et al. [67] showed that heterozygous Tsc2 mice had abnormalities in hippocampal long-term potentiation, with induction of late long-term potentiation at a lower threshold. This led to deficits in spatial learning and contextual discrimination, even in the absence of frank neuropathology and seizures, and these deficits were abrogated with only 5 days of rapamycin treatment. Various approaches have been used to investigate the underlying mechanisms responsible for this hippocampal hyperexcitability [32], with at least 2 contributing pathways implicated to date. Tavazoie et al. [28] reported that loss of Tsc2 is associated with increased α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid(AMPA)-mediated excitatory currents, whereas Chevere-Torres et al. [26] found that metabotropic glutamate-induced long-term depression is impaired with functional disruption of Tsc2 (in the DeltaRG mouse model of TSC) that is extracellular regulated kinase dependent and rapamycin sensitive [27]. This hyperexcitability is thought to contribute to the epilepsy and learning problems seen in TSC and demonstrates another mechanism in which mTOR inhibitors may prove beneficial for the treatment of ASD.
Several studies have specifically examined the underlying mechanisms of autism-like behaviors in animals with a TSC mutation. In contrast to hippocampal neurons, where loss of Tsc1 or Tsc2 results in hyperexcitability, loss of Tsc1 in cerebellar Purkinje cells in mice results in decreased neuronal excitability [30]. These latter mice demonstrate an ASD-like phenotype, including abnormal social behavior and learning, repetitive behaviors, and vocalization. These changes were prevented by treatment with rapamycin. Similar results were obtained with Tsc2 knockout in the Purkinje neurons [57]. Talos et al. [72] reported that a rodent model of neonatal seizures results in mTORC1 overactivity, and seizures during a period of peak synaptogenesis contributed to deficits in a social novelty assay. Treatment with rapamycin in animals with neonatal hypoxic seizures was protective against the development of epilepsy and decreased the social novelty deficit. Another mouse model with conditional knockout of Tsc1 in forebrain neurons (under CaMKIIα-cre) further links mTOR hyperactivation with seizures and autism-like behaviors [73]. Decreased social behavior (in the 3-chamber social approach) and increased repetitive behaviors (marble burying) were observed in these mutant mice. As the recurrent seizure spread to the brainstem, the authors hypothesized that serotonergic neurons were specifically implicated in mediating the autistic-like behaviors. Using a conditional knockout of Tsc1 in serotonergic neurons, they found that autism-like behaviors persisted, even though the mice did not develop spontaneous seizures. Treatment with rapamycin decreased mTOR hyperactivity and normalized measures of social preference and marble burying. Taken together, these studies show a complex relationship between early brain development, mTOR pathway overactivity in various brain regions, seizures, and the development of ASD-like behaviors, and also show that treatment with an mTOR inhibitor is a potentially promising method to normalize underlying mechanisms leading to the behavioral manifestations of ASD.
Preliminary Results of Neuropsychological Effects of mTOR Inhibitor Trials in Humans
The first human clinical trials using mTOR inhibitors in the treatment of TSC began in 2002, with the initial focus on reducing tumor burden and growth in the brain (SEGA), kidney (angiomyolipoma), and lung (lymphangioleiomyomatosis). These targets were selected based on significant disease-related morbidity and mortality, lack of effective noninvasive treatment options, and relative ease in objectively assessing treatment response. Rapamycin was shown to be effective and well tolerated for reducing SEGA tumor volume [74], renal angiomyolipoma volume [75–77], and pulmonary lung function [78], as long as treatment was maintained. However, in each case disease progression resumed upon discontinuation of treatment, similar to what had been observed in preclinical models. As a result, more recent studies have evaluated the sustained efficacy and long-term safety of continuous treatment. Krueger et al. [79, 80] treated 28 patients with SEGAs with everolimus for a median of 3 years without loss of SEGA tumor volume reduction and no newly encountered clinical toxicities. A placebo-controlled SEGA treatment study with everolimus involving 117 patients, with a median follow-up of 2 years, yielded similar results [81, 82]. Bissler et al. [83] reported safety and efficacy of everolimus treatment for angiomyolipoma but median treatment duration was much shorter (slightly over 6 months).
Throughout these early clinical trials, unpublished anecdotal reports (Franz, NIH Curing Epilepsy 2007), case reports [84], and secondary end points [77, 79] suggested that rapalogs might have benefit for central nervous system (CNS)-related manifestations in TSC beyond reduction of tumors and tubers. For example, Krueger et al. [79] reported after 6 months of treatment with everolimus, seizure frequency was reduced in 56 % of participants. By 24 months, patients with daily seizures were reduced from 27 % to 13 %, while patients with no seizures increased from 39 % to 65 % during the same interval [80]. These improvements correlated with improvement in white matter integrity as measured with diffusion tensor imaging and were independent of SEGA treatment response [85]. Analysis of seizure control as a secondary end point in the placebo-controlled, blinded Phase III trial with everolimus to treat SEGA was unable to confirm these earlier results, largely owing to significant differences in baseline seizure frequency between the control and treatment groups, and the fact that in both groups the majority of participants were seizure-free at time of treatment initiation [81]. Since then, smaller, mostly retrospective, studies have continued to report treatment benefit for epilepsy [86–89]. To date, the only clinical trial specifically to use mTOR inhibitors to treat epilepsy prospectively involved open-label treatment with everolimus for 4 months [90]. Eighteen of 20 participants reported a reduction in seizure frequency, with 12/20 (60 %) experiencing an improvement of 50 % or more. Furthermore, treatment response appeared to improve over time, with better response at 3–4 months compared with response at 1–2 months. This delayed or prolonged response time for optimal outcome supports involvement of mechanisms discussed in the previous sections that require weeks to months rather than hours to days to show effect, such as synapse remodeling, network connectivity, and neuronal plasticity. However, direct evidence that this is the case and the relative contributions each has on seizure susceptibility and treatment response remains to be determined. Longer-term treatment and placebo-controlled, double-blind clinical trials are already in progress to further investigate everolimus impact on seizures and epilepsy in TSC.
Evaluating the impact of mTOR inhibitors in patients with TSC with intellectual disability and other aspects of TAND, including ASD, has been more difficult. In the initial 28-patient SEGA study, a robust neurocognitive assessment battery was included but the majority of participants were developmentally and/or cognitively impaired such that relatively few were able to complete the assessments, and no conclusions could be drawn [79]. A similar attempt by Davies et al. [77] in adults treated with rapamycin for angiomyolipoma fared a little better, although the final size of the analysis cohort remained relatively small (n = 8) and yielded mixed results. They reported improvements in recall memory (7/8) and executive function (5/8) but worsening in recognition memory (5/8) in adults with TSC following treatment with rapamycin for 4–12 months. In another study, Chung et al. [91] reported improvement in 3 patients with TSC with comorbid intermittent explosive disorder or adjustment disorder not otherwise specified who underwent formal psychiatric evaluations before and after initiating treatment with everolimus (n = 2) or rapamycin (n = 1). In the more recent open-label epilepsy trial by Krueger et al. [90], an indirect, more broad approach was utilized, sacrificing direct observational measures for indirect parental report using validated assessment tools with the goal of providing more universal assessment of TAND-related comorbidities and domains. Using the Nisonger Child Behavior Rating Form, adapted from the Child Behavioral Rating Form to allow assessment of both cognitively impaired and normal IQ children, they reported small but significant improvement in adaptive social behaviors, conduct problems, and insecurity/anxiety compared with baseline following treatment for 4 months. Quality of Life for Children with Epilepsy assessments done in parallel over the same time period identified similar improvements in multiple domains, including attention and concentration, behavior, social interactions and activity, and overall quality of life. However, whether these changes were secondary to or independent of improvement in seizure control could not be determined.
Intense interest exists to confirm and expand these preliminary studies in order to determine if a longer treatment period results in sustained or further improvements, to verify results in a larger cohort using a placebo-controlled, double-blinded study design, and to incorporate direct observational assessment approaches to better determine the specific areas of neurocognitive and ASD-related subdomains affected by mTOR inhibitor treatment and further separate which effects are seizure control-dependent and which are seizure control-independent. Ongoing trials include Everolimus (RAD001) Therapy for Epilepsy in Patients with TSC (clinicaltrials.gov: NCT01070316); Trial of RAD001 and Neurocognition in TSC (clinicaltrials.gov: NCT01289912); Efficacy of RAD001/Everolimus in Autism and NeuroPsychological Deficits in Children With Tuberous Sclerosis Complex (RAPIT) (clinicaltrials.gov: NCT01730209); A Placebo-controlled Study of Efficacy & Safety of 2 Trough-ranges of Everolimus as Adjunctive Therapy in Patients With TSC & Refractory Partial-onset Seizures (EXIST-3) (clinicaltrials.gov: NCT01713946); A Study of Everolimus in the Treatment of Neurocognitive Problems in Tuberous Sclerosis (TRON) (clinicaltrials.gov: NCT01954693); Rapalogues for Autism Phenotype in TSC: A Feasibility Study (RAPT) (clinicaltrials.gov: NCT01929642); and Long-term Follow-up for Growth and Development of Pediatric Patients From CRAD001M2301 (EXIST-LT) (clinicaltrials.gov: NCT02338609).
Early Vigabatrin Treatment Trials
Vigabatrin is a rationally designed drug that aims to increase the levels of GABA, the main inhibitory neurotransmitter in the CNS. The addition of a vinyl group to GABA (vi-GABA-trin) creates a substrate that irreversibly inhibits GABA-transaminase, the GABA-degrading enzyme, thus increasing GABA availability in the synaptic cleft. In humans, it has a specific and rapid mechanism of action and its antiepileptic properties are thought to be a direct result of increased inhibitory neurotransmission, although additional mechanisms are considered [92].
Side effects during initiation are those common to drugs affecting the CNS, and include irritability, drowsiness, and hypotonia, which typically resolve over time. One important side effect is irreversible peripheral visual field constriction, with a risk that increases cumulatively with higher doses and longer exposure [93]. In the USA, the manufacturer has implemented a vigorous monitoring program for this side effect. The clinically evident response of cessation of spasms within days to weeks provides an opportunity to try vigabatrin at a low risk.
Other than as add-on for intractable complex partial seizures, vigabatrin is approved in the US as monotherapy for infantile spasms (IS). An early small controlled trial demonstrated efficacy compared with placebo but no patients with TSC were included [94]. In studies of high- versus low-dose vigabatrin and of vigabatrin versus steroid therapy for IS, patients with TSC had consistently higher response rates to vigabatrin [95, 96]. Indeed, an early review of the literature suggested a response rate of 95 % to vigabatrin in IS due to TSC [97], an estimate that has not changed in subsequent literature [93]. Within patients with TSC, in a head-to-head comparison with adrenocorticotropic hormone, vigabatrin showed clear superiority [98]. Based on these and other studies and reviews, the International TSC Consensus Conference recommends vigabatrin as first-line treatment for IS in TSC [4].
Why IS in TSC in particular respond so dramatically to vigabatrin is not understood. Other drugs affecting GABAergic transmission (e.g., barbiturates and benzodiazepines) do not achieve comparable efficacy. Reported additional effects of vigabatrin such as decreased glial GABA uptake, enhanced GABA release, and reduction of glutamate have not been reproduced [92]. In a TSC mouse model, vigabatrin suppressed the mTOR pathway, which could be an additional explanation for the particular efficacy in TSC [70]. However, the rapid efficacy against IS is more likely based on GABAergic changes.
Better long-term neurocognitive outcome has been reported with early, aggressive, and successful treatment of IS [99–101], and later cessation of IS is associated with poorer outcomes [102]. For partial seizures, however, efficacy is lower and the beneficial effects on outcome may not be present when compared with treatment of IS [100, 101].
When given even earlier, in a small open-label trial of vigabatrin initiation prior to onset of IS, good long-term outcomes were also seen [46]. If, indeed, mTOR pathway activity is modified by vigabatrin, it could contribute to these longer-term benefits. This raises the possibility that vigabatrin is not merely antiepileptic, but it may also be disease modifying.
How to identify who would most benefit, with justifiable risk, from early or even pre-emptive intervention with vigabatrin, is a subject of active study. For example, epileptic spasms beyond infancy respond less well to vigabatrin, even though these seizures are similar to IS in their electroclinical presentation. Thus, there appears to be a therapeutic window in obtaining the longer-term neurodevelopmental benefits from early medical intervention. In the USA, a multicenter prospective observational study is currently underway to identify early developmental, neurophysiology, and neuroimaging markers for increased risk of epilepsy and ASD in TSC (clinicaltrials.gov: NCT01767779; NCT01780441). In a next phase, those markers will be used to stratify patients to either standard of care versus early aggressive, pre-emptive treatment with vigabatrin. In Europe, a similar effort is ongoing (clinicaltrials.gov: NCT02098759).
Future Directions of Treatment for ASD in TSC
We are on the cusp of a new era in targeted, disease-modifying treatment for TSC and other disorders associated with ASD. The initial results from a Phase II study looking specifically at effects on neurocognition in TSC after treatment with everolimus (clinicaltrials.gov: NCT01289912) are currently in the process of being analyzed. Open questions include the type of effects mTOR inhibitors will have on ASD symptoms in TSC and how the effects will vary based on patient characteristics and disease manifestations. Another crucial question is what end points are quantifiable and dynamic in response to treatment within the duration of the trial. Finally, it is not yet clear whether there is a critical window during which treatments will be most effective. Once these questions are studied, we will be in a better position to determine which patients will most benefit from which treatment.
Robust and early biomarkers of neurodevelopmental outcome in TSC are critical to best target treatments to patients who will most benefit from them, to avoid unnecessarily exposing patients to potentially harmful and irreversible treatment side effects, and to measure treatment effects. For ASD, behavioral and developmental signs may be recognized early in high-risk populations [10], and retrospective work has identified electroencephalography network properties and diffusion tensor imaging metrics as possible biomarkers [39, 103]. Diffusion imaging abnormalities may reflect changes in the underlying neurobiology [43], and these measures may even respond to intervention in parallel with clinical changes seen [85]. Further research on these and other biomarkers is needed to better characterize their utility for clinical use and future prospective studies.
It is a rare but fortunate occurrence when our understanding of a disorder matches our ability to treat based on that understanding. We are approaching that point now for ASD in TSC. Our hope is that the recent advances described here in understanding and treating the mechanisms contributing to ASD in TSC will benefit our patients with TSC and provide a path towards better understanding and treatment for all people with ASD.
References
Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci 1991;615:125-127.
Curatolo P, Bombardieri R, Jóźwiak S. Tuberous sclerosis. Lancet 2008;372:657-668.
Curatolo P, Maria BL. Tuberous sclerosis. Handb Clin Neurol 2013;111:323-331.
Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:255-265.
de Vries PJ, Whittemore VH, Leclezio L, et al., Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol 2015;52:25-35.
Jeste SS, Sahin M, Bolton P, Ploubidis GB, Humphrey A. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol 2008;23:520-525.
Leclezio L, de Vries PJ. Advances in the treatment of tuberous sclerosis complex. Curr Opin Psychiatry 2015;28:113-120.
Ehninger D, Sano Y, de Vries PJ, et al., Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol Psychiatry 2012;17:62-70.
Datta AN, Hahn CD, Sahin M. Clinical presentation and diagnosis of tuberous sclerosis complex in infancy. J Child Neurol 2008;23:268-273.
Spurling Jeste S, Wu JY, Senturk D, et al. Early developmental trajectories associated with ASD in infants with tuberous sclerosis complex. Neurology 2014;83:160-168.
Bruining H, Eijkemans MJ, Kas MJ, et al. Behavioral signatures related to genetic disorders in autism. Mol Autism 2014;5:11.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274-293.
Lipton JO, Sahin M. The neurology of mTOR. Neuron 2014;84:275-291.
Ehninger D. From genes to cognition in tuberous sclerosis: implications for mTOR inhibitor-based treatment approaches. Neuropharmacology 2013;68: 97-105.
Feliciano DM, Lin TV, Hartman NW, et al., A circuitry and biochemical basis for tuberous sclerosis symptoms: from epilepsy to neurocognitive deficits. Int J Dev Neurosci 2013;31:667-678.
Chen J, Alberts I, Li X. Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. Int J Dev Neurosci 2014;35:35-41.
Tang G, Gudsnuk K, Kuo SH, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014;83:1131-1143.
Di Nardo A, Wertz MH, Kwiatkowski E, et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum Mol Genet 2014;23:3865-3874.
Di Nardo A, Kramvis I, Cho N, et al. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci 2009;29:5926-5937.
Lim K-C, Crino PB. Focal malformations of cortical development: new vistas for molecular pathogenesis. Neuroscience 2013;252:262-276.
Hoeffer CA, Klann E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci 2010;33:67-75.
de Vries PJ. Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics 2010;7:275-282.
Kelleher RJ, Bear MF. The autistic neuron: troubled translation? Cell 2008;13:401-406.
Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011;480:63-68.
Won H, Mah W, Kim E. Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front Mol Neurosci 2013;6:19.
Chevere-Torres I, Kaphzan H, Bhattacharya A, et al. Metabotropic glutamate receptor-dependent long-term depression is impaired due to elevated ERK signaling in the DeltaRG mouse model of tuberous sclerosis complex. Neurobiol Dis 2012;45:1101-1110.
Hou, L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci 2004;24:6352-6361.
Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 2005;8:1727-1734.
Santini E, Klann E. Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal 2014;7:re10.
Tsai PT, Hull C, Chu Y, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012;488:647-651.
Zeng L-H, Ouyang Y, Gazit V, et al. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis 2007;28:184-196.
Bateup HS, Johnson CA, Denefrio CL, et al. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 2013;78:510-522.
Curatolo P. Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr Neurol 2014;52:281-289.
Crino PB. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol 2013;125:317-332.
Bolton P, Park RJ, Higgins J, Griffiths PD, Pickles A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain 2002;125:1247-1255.
Eluvathingal TJ, Behen ME, Chugani HT, et al. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol 2006;21:846-851.
Weber AM, Egelhoff JC, McKellop JM, Franz DN. Autism and the cerebellum: evidence from tuberous sclerosis. J Autism Dev Disord 2000;30:511-517.
Meikle L, Talos DM, Onda H, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 2007;27:5546-5558.
Peters JM, Sahin M, Vogel-Farley VK, et al. Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad Radiol 2012;19:17-25.
Lewis WW, Sahin M, Scherrer B, et al. Impaired language pathways in tuberous sclerosis complex patients with autism spectrum disorders. Cereb Cortex 2013;23:1526-1532.
Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol 2007;17:103-111.
Wass S. Distortions and disconnections: disrupted brain connectivity in autism. Brain Cogn 2011;75:18-28.
Peters JM, Taquet M, Prohl AK, et al. Diffusion tensor imaging and related techniques in tuberous sclerosis complex: review and future directions. Future Neurol 2013;8:583-597.
Waltereit R, Japs B, Schneider M, de Vries PJ, Bartsch D. Epilepsy and Tsc2 haploinsufficiency lead to autistic-like social deficit behaviors in rats. Behav Genet 2011;41:364-372.
Jansen FE, Vincken KL, Algra A, et al. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology 2008;70:916-923.
Jóźwiak S, Kotulska K, Domańska-Pakieła D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol 2011;15:424-431.
Domanska-Pakiela D, Kaczorowska M, Jurkiewicz E, et al. EEG abnormalities preceding the epilepsy onset in tuberous sclerosis complex patients—a prospective study of 5 patients. Eur J Paediatr Neurol 2014;18:458-468.
van Eeghen AM, Pulsifer MB, Merker VL, et al. Understanding relationships between autism, intelligence, and epilepsy: a cross-disorder approach. Dev Med Child Neurol 2013;55:146-153.
Berg AT, Plioplys S. Epilepsy and autism: is there a special relationship? Epilepsy Behav 2012;23:193-198.
El Achkar CM, Spence SJ. Clinical characteristics of children and young adults with co-occurring autism spectrum disorder and epilepsy. Epilepsy Behav 2015. doi:10.1016/j.yebeh.2014.12.022
Careaga M, Van de Water J, Ashwood P. Immune dysfunction in autism: a pathway to treatment. Neurotherapeutics 2010;7:283-292.
Gentzler RD, Altman JK, Platanias LC. An overview of the mTOR pathway as a target in cancer therapy. Expert Opin Ther Targets 2012;16:481-489.
Santulli G, Totary-Jain H. Tailoring mTOR-based therapy: molecular evidence and clinical challenges. Pharmacogenomics 2013;14:1517-1526.
Lebrun-Julien F, Bachmann L, Norrmen C, et al. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J Neurosci 2014;34:8432-8448.
Normand EA, Crandall SR, Thorn CA, et al. Temporal and mosaic Tsc1 deletion in the developing thalamus disrupts thalamocortical circuitry, neural function, and behavior. Neuron 2013;78:895-909.
Mietzsch U, McKenna J, 3rd, Reith RM, Way SW, Gambello MJ. Comparative analysis of Tsc1 and Tsc2 single and double radial glial cell mutants. J Comp Neurol 2013;521:3817-3831.
Reith RM, McKenna J, Wu H, et al. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol Dis 2013;51:93-103.
Magri L, Cominelli M, Cambiaghi M, et al. Timing of mTOR activation affects tuberous sclerosis complex neuropathology in mouse models. Dis Model Mech 2013;6:1185-1197.
Fu C, Cawthon B, Clinkscales W, et al. GABAergic interneuron development and function is modulated by the Tsc1 gene. Cereb Cortex 2012;22:2111-2119.
Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol Dis 2012;45:369-380.
Yuan E, Tsai PT, Greene-Colozzi E, et al. Graded loss of tuberin in an allelic series of brain models of TSC correlates with survival, and biochemical, histological and behavioral features. Hum Mol Genet 2012;21:4286-4300.
Magri L, Cambiaghi M, Cominelli M, et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 2011;9:447-462.
Zhou J, Shrikhande G, Xu J, et al. Tsc1 mutant neural stem/progenitor cells exhibit migration deficits and give rise to subependymal lesions in the lateral ventricle. Genes Dev 2011;25:1595-1600.
Goto J, Talos DM, Klein P, et al. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci U S A 2011;108:E1070-E1079.
Zeng LH, Rensing NR, Zhang B, et al. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet 2011;20:445-454.
Meikle L, Pollizzi K, Egnor A, et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 2008;28:5422-5432.
Ehninger D, Han S, Shilyansky C, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med 2008;14:843-848.
Way SW, McKenna J, 3rd, Mietzsch U, et al. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum Mol Genet 2009;18:1252-1265.
Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 2008;63:444-453.
Zhang B, McDaniel SS, Rensing NR, Wong M. Vigabatrin inhibits seizures and mTOR pathway activation in a mouse model of tuberous sclerosis complex. PLoS ONE 2013;8:e57445.
Uhlmann EJ, Wong M, Baldwin RL, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol 2002;52:285-296.
Talos DM, Sun H, Zhou X, et al. The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS ONE 2012;7:e35885.
McMahon JJ, Yu W, Yang J, et al. Seizure-dependent mTOR activation in 5-HT neurons promotes autism-like behaviors in mice. Neurobiol Dis 2015;73:296-306.
Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 2006;59:490-498.
Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 2008;358:140-151.
Dabora SL, Franz DN, Ashwal S, et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF- D levels decrease. PLoS ONE 2011;6:e23379.
Davies DM, de Vries PJ, Johnson SR, et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res 2011;17:4071-4081.
McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 2011;364:1595-1606.
Krueger DA, Care MM, Holland K et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 2010;363:1801-1811.
Krueger DA, Care MM, Agricola K, et al. Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology 2013;80:574-580.
Franz, D.N., E. Belousova, S. Sparagana, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013;381:125-132.
Franz DN, Belousova E, Sparagana S, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol 2014;15:1513-1520.
Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013;381:817-824.
Muncy J, Butler IJ, Koenig MK. Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol 2009;24:477.
Tillema J-M, Leach JL, Krueger DA, Franz DN. Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology 2012;78:526-531.
Kotulska K, Chmielewski D, Borkowska J, et al. Long-term effect of everolimus on epilepsy and growth in children under 3 years of age treated for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Eur J Paediatr Neurol 2013;17:479-485.
Wiegand G, May TW, Ostertag P, et al. Everolimus in tuberous sclerosis patients with intractable epilepsy: a treatment option? Eur J Paediatr Neurol 2013;17:631-638.
Canpolat M, Per H, Gumus H, et al. Rapamycin has a beneficial effect on controlling epilepsy in children with tuberous sclerosis complex: results of 7 children from a cohort of 86. Childs Nerv Syst 2014;30:227-240.
Cardamone M, Flanagan D, Mowat D, et al. Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr 2014;164:1195-1200.
Krueger DA, Wilfong AA, Holland Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol 2013;74:679-687.
Chung TK, Lynch ER, Fiser CJ, et al. Psychiatric comorbidity and treatment response in patients with tuberous sclerosis complex. Ann Clin Psychiatry 2011;23:263-269.
Ben-Menachem E. Mechanism of action of vigabatrin: correcting misperceptions. Acta Neurol Scand Suppl 2011:5-15.
Willmore LJ, Abelson MB, Ben-Menachem E, Pellock JM, Shields WD. Vigabatrin: 2008 update. Epilepsia 2009;50:163-173.
Appleton RE, Peters AC, Mumford JP, Shaw DE. Randomised, placebo-controlled study of vigabatrin as first-line treatment of infantile spasms. Epilepsia 1999;40:1627-1633.
Elterman RD, Shields WD, Mansfield KA, Nakagawa J; US Infantile Spasms Vigabatrin Study Group, Randomized trial of vigabatrin in patients with infantile spasms. Neurology 2001;57:1416-1421.
Vigevano F, Cilio MR. Vigabatrin versus ACTH as first-line treatment for infantile spasms: a randomized, prospective study. Epilepsia 1997;38:1270-1274.
Hancock E, Osborne JP. Vigabatrin in the treatment of infantile spasms in tuberous sclerosis: literature review. J Child Neurol 1999;14:71-74.
Chiron C, Dumas C, Jambaque I, Mumford J, Dulac O. Randomized trial comparing vigabatrin and hydrocortisone in infantile spasms due to tuberous sclerosis. Epilepsy Res 1997;26:389-395.
Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol 2010;14:146-149.
Jambaque I, Chiron C, Dumas C, Mumford J, Dulac O. Mental and behavioural outcome of infantile epilepsy treated by vigabatrin in tuberous sclerosis patients. Epilepsy Res 2000;38:151-160.
Yum MS, Lee EH, Ko TS. Vigabatrin and mental retardation in tuberous sclerosis: infantile spasms versus focal seizures. J Child Neurol 2013;28:308-313.
Muzykewicz DA, Costello DJ,. Halpern EF, Thiele EA. Infantile spasms in tuberous sclerosis complex: prognostic utility of EEG. Epilepsia 2009;50:290-296.
Peters JM, Taquet M, Vega C, et al. Brain functional networks in syndromic and non-syndromic autism: a graph theoretical study of EEG connectivity. BMC Med 2013;11:54.
Acknowledgments
P.E.D. is supported by a training grant from the National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke (NINDS; 3R25NS070682-04S1). J.M.P., D.A.K. and M.S. are supported by NIH (U01 NS082320, P20 NS080199, U54NS092090). The Developmental Synaptopathies Consortium (U54NS092090) is a part of the National Center for Advancing Translational Sciences (NCATS) Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through collaboration between NCATS, National Institute of Mental Health, NINDS and National Institute of Child Health and Human Development. J.M.P. is also supported by Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, NIH Award UL1 TR001102). D.A.K. is also supported by Tuberous Sclerosis Alliance, Clack Foundation, Novartis, and Upsher-Smith. Research in the laboratory of M.S. is also supported by the NIH P30 HD018655, the Department of Defense, Tuberous Sclerosis Alliance, Autism Speaks, Nancy Lurie Marks Family Foundation, Simons Foundation, Boston Children’s Hospital Translational Research Program, and Novartis, Shire and Roche. Full conflict of interest disclosures are available in the electronic supplementary material for this article. Owing to limited space, we have not quoted all the literature in this field, and we apologize to those whose articles are not referenced. Finally, we are indebted to the children and families who participated in the studies reviewed in this article.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 498 kb)
Rights and permissions
About this article

Cite this article
Davis, P.E., Peters, J.M., Krueger, D.A. et al. Tuberous Sclerosis: A New Frontier in Targeted Treatment of Autism. Neurotherapeutics 12, 572–583 (2015). https://doi.org/10.1007/s13311-015-0359-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-015-0359-5