
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease leading to cell death of predominantly motor neurons. Despite extensive research in this disease, finding a way to slow the progress of the disease has been challenging. RNA-targeted therapeutic approaches, including small interfering RNA and antisense oligonucleotides are being developed for genetic forms of ALS. ALS provides an unique opportunity for the use of RNA inhibition strategies given a well-defined animal model, extensive available information regarding the causative genes, and recent experience in phase 1 clinical trial.
Similar content being viewed by others

With US Food and Drug Administration approval for Mipomersen for familial hypercholesterolemia [1], and the recent successful phase 2a trial for Miravirsen in patients with hepatitis C [2], RNA-targeted therapeutics have entered a new phase of growth. Neurodegenerative disorders provide immense potential for such genetic manipulation given the role of disruption of RNA and protein metabolism in pathogenesis of multiple disorders such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, spinocerebellar ataxias, muscular dystrophy, and amyotrophic lateral sclerosis (ALS) [3, 4]. With antisense oligonucleotides (ASOs) now moving towards a phase 3 trial in spinal muscular atrophy [5], this mode of therapy may hold the key for these previously incurable neurological illnesses [6].
ALS, or Lou Gehrig’s disease, a progressive upper and lower motor neuron degenerative disorder, occurs at one end of a spectrum of disorders that cause frontotemporal dementia at the other end [7]. While most of the sporadic cases of the disorder do not have a known genetic basis, a majority of the familial forms have now been identified to have a genetic basis with mutations in several genes such as SOD1, TARDBP, FUS, and C9ORF72 [8]. Even in the absence of mutations, rare variants in these genes have been shown to be associated with both familial and sporadic forms of ALS [9]. These mutations typically cause protein aggregates and a gain-of-toxic function effect that lead to cell-specific degeneration [10]. While medications to significantly reduce this gain-of-toxic function effect have not been discovered, targeting the production of toxic protein or RNA could achieve this aim.
RNA-targeted therapies include ASOs and small interfering RNA (siRNA), both of which utilize Watson–Crick base pairing-focused antisense mechanisms that cause degradation of or inactivate a particular mRNA [11]. These separate pathways are distinguished by the enzymes that degrade the RNA, by their sites of action, and by the different methods employed to use siRNA or ASOs therapeutically. RNA interference (RNAi) is an endogenous cell physiologic mechanism to regulate mRNA at the level of post-transcriptional processing in the cytoplasm via the RNA-induced silencing complex [12]. The Agonaute 2 in RNA-induced silencing complex has endonuclease activity and degrades the target mRNA [11]. Drs Andrew Fire and Craig Mello received the Nobel Prize in Medicine in 2006 for discovering RNAi in Caenorhabditis elegans [13]. A more robust pathway to reduce the levels of target RNA involves the use of double-stranded siRNA that activates this same pathway, and leads to mRNA cleavage and more potent inhibition of target mRNA [14]. However, this potent inhibition is offset by a relatively short half-life [15]. The siRNA can be continually produced from a viral vector [short hairpin RNA (shRNA)] and thus delivered to animal models or potentially to humans. Efforts are also underway to chemically stabilize siRNA, which will avoid the need for viral vectors [16]. ASOs are chemically modified, single-stranded oligonucleotides that activate RNAse H, an endonuclease in the nucleus, to degrade the cognate mRNA that binds to the ASOs. These also arrest mRNA translation in the cytoplasm and can alter mRNA splicing [17]. Unlike the relatively newly recognized endogenous RNAi pathways, the cellular response to degrade double-stranded RNA has been recognized for decades, and there has been extensive research to determine chemical modifications to ASOs to increase the stability in biological fluids and increase the potency without activating an immune response. ASOs for use outside of the nervous system have been developed for multiple diseases and recently resulted in an US Food and Drug Administration-approved drug for familial hypercholesterolemia (mipomersen) [18]. The promise of using ASOs to treat neurodegenerative diseases has been greatly bolstered by recent demonstrations of wide spread distribution of ASOs throughout the brain and spinal cord when delivered to the cerebrospinal fluid (CSF) [19–21].
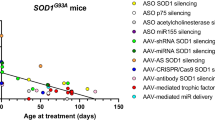
The murine ALS G93A model overexpresses mutant human superoxide dismustase 1 (SOD1) and develops progressive motor neuron disease [22]. This model has been well characterized and provides a defined target for RNAi studies. The concept of RNAi for decreasing mutant SOD1 received validation in vitro with successful reduction in mutant human SOD1 in cell lines transfected with mutant SOD1 in the presence of siRNA or shRNA against SOD1 [23]. The first successful in vivo RNAi studies in the ALS mouse model used adeno-associated virus (AAV) or lentiviral transduction of shRNA against human SOD1. Miller et al. [24] demonstrated increased strength in SOD1G93A mice by intramuscular delivery and retrograde transport of AAV-2–shRNA SOD1. Intraspinal injections of lentivirus with shRNA SOD1 [25], or intramuscular injections of lentivirus with shRNA SOD1 and rabies envelop protein to increase retrograde transport led to delayed onset of motor neuron disease in SOD1G93A mice. Ralph et al. [25] showed a 115% delay in onset of disease and a 77% increase in survival, while Raoul et al. [26] showed a 20% delay in onset of disease. Each of these 3 early studies was limited by the viral methods available. Subsequent studies using more potent shRNA and AAV-9 with a more widespread delivery of shRNA showed greater reduction of SOD1 and delivery with a paradigm that has greater potential for translation forward to the clinic. Recently, peripheral, as well as intrathecal, injection of an AAV encoding shRNA against human SOD1 mRNA has also been shown to delay disease onset and progression in G93A mice by about 30–40% [27, 28].
Smith et al. [21] demonstrated, for the first time, that CSF delivery of ASOs resulted in widespread delivery of ASOs to the brain and spinal cord. Using this CSF delivery paradigm in SOD1G93A rats at onset, they demonstrated a slowing of disease progression by about 37%. These animal studies led to a successful phase 1 study in humans of intrathecally delivered ASOs against SOD1 that was well tolerated and had no significant adverse events [29]. They also established important first-in-human CSF pharmacokinetics for ASOs that will greatly aid in future ASO clinical trials. As CSF SOD1 can be an excellent marker with which to look at the effectiveness of ASO treatment [30], this therapy can be guided and monitored by measuring CSF SOD1 levels. While a role for misfolded wild-type SOD1 in sporadic ALS remains inconclusive, some evidence for such a role exists through cell culture models and use of confirmation-specific antibodies that are able to distinguish misfolded SOD1 from normal SOD1 in human autopsy tissues [31–36]. If ASOs against SOD1 continue to be safe, these have the potential to be used in cases of sporadic ALS where they could reduce the amount of misfolded SOD1. Development of an assay that could demonstrate misfolded SOD1 involvement in ALS in an individual living patient would greatly strengthen the rationale for applying SOD1-based therapeutics to non-SOD1 mutation-carrying ALS.
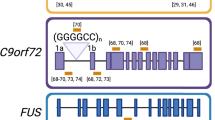
Mutations in the form of increased hexanucleotide repeat expansions in C9ORF72 are a major cause of familial ALS and are also found in a significant minority of patients with sporadic ALS [8]. Although there are no murine models of this mutation, fibroblasts derived from patients with C9ORF72 mutations can be manipulated to provide a human stem cell-derived neuronal model of ALS/frontotemporal dementia [37]. These cultured C9ORF72 neuronal cells show RNA foci and increased susceptibility to excitotoxicity [37–39]. Use of ASOs against the hexanucleotide repeats leads to decreased RNA foci in these neurons and decreased vulnerability to excitotoxicity [37–39]. Given the lack of an animal model of C9ORF72, these ASOs have not been tested further in vivo but hold promise that they may be able to reverse pathology in C9ORF72 patients.
An alternative approach to reducing the known aberrant mRNA directly is to use RNAi against a downstream pathway that may have a similar effect of slowing the neurodegenerative process. Such an approach has the advantage that it can be used even in cases where the mutation causing the disease is not known, as in the vast majority of patients with spontaneous ALS [8]. One such approach was shown early to have such an effect. Mutant SOD1 motor neurons had been shown to have increased sensitivity to Fas activation, leading to increased cell death by apoptotic pathways [40]. Intrathecal injection of siRNA against Fas was then shown to delay disease onset (by 20%), improve motor function and prolong survival (by 14%) . Yet another approach is to target miRNAs with ASOs such that negative regulation of the mRNA can be reversed. Such an approach has been used for silencing miR-122 in hepatocytes, thereby shutting down propagation of hepatitis C viral particles [2]. This approach has also been shown to be effective in G93A mice. MiR-155 was shown to be elevated along with 5 other miRs in human and mice ALS tissues [41]. An ASO generated against miR-155 was then shown to prolong survival (by 7%) and prolong disease duration (by 38%) when injected intraventricularly after disease onset [41]. Similar slowing of disease in SOD1G93A mice and prolongation of survival (by 8%) was achieved by a second group inhibiting miR-155 [42].
These findings in ALS and similar such findings in spinal muscular atrophy, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, spinocerebellar ataxias, and other neurodegerative disorders thus provide promise that RNA targeted therapeutics may be a way to control these progressive disorders [43]. Several issues regarding such genetic manipulation in humans still need to be addressed. First, while the first ASO clinical trial in the central nervous system (CNS) shows great promise from a safety/tolerability point of view [29], longer-term data in the next studies are needed to more fully assess tolerability. Whereas earlier ASOs tended to elicit immune responses, modifications in these ASOs now are better tolerated [18]. Second, an important next step will be to show that the RNA-targeted therapeutic does, indeed, affect the target mRNA/protein in the CNS. The doses given by Miller et al. [29] were insufficient to lower SOD1 in their phase 1 trial. Third, for viral delivery the inability to “turn off” the virally delivered shRNA remains a concern that continues to be the topic of research. ASOs will be cleared after delivery, but the long half-life (30–60 days) of the ASOs in the CNS may present a similar concern for how to reverse any unwanted side effects [29]. Last, it remains unknown whether lowering levels of the causative gene (e.g., SOD1) in ALS in symptomatic patients will be soon enough to reverse or slow the disease process. Treating presymptomatically, as is being done in Alzheimer’s disease trials, though feasible clinically, would present an enormous challenge for an SOD1 ALS clinical trial given the low numbers of patients and the high variability in age of onset [44]. Despite these concerns, RNA-targeted therapies show immense promise for neurodegenerative syndromes and raise hope for a therapy that may be able to slow ALS disease progression.
References
Hair P, Cameron F, McKeage K. Mipomersen sodium: first global approval. Drugs 2013;73:487-493.
Janssen HL, Reesink HW, Lawitz EJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med 2013;368:1685-1694.
Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013;154:727-736.
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med 2004;10(Suppl.):S10-S17.
Finkel R, Day J, Chiriboga CC, et al. Results of a phase 2 open-label study of ISIS-SMNRx in Patients with infantile (type 1) spinal muscular atrophy. AAN Abstracts 2014;Neurology 2014;82(10 Supplement):S6.003
DeVos SL, Miller TM. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics 2013;10:486-497.
Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013;14:248-264.
Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17:17-23.
Cady J, Allred P, Bali T, et al. ALS onset is influenced by the burden of rare variants in known ALS genes. Ann Neurol 2015;77:100-113.
Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 2011;7:616-630.
Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol 2012;19:60-71.
Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell 2009;136:642-655.
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806-811.
Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet 2007;8:173-184.
Deng Y, Wang CC, Choy KW, et al. Therapeutic potentials of gene silencing by RNA interference: principles, challenges, and new strategies. Gene 2014;538:217-227.
Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature 2009;457:426-433.
Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 2012;11:125-140.
Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol 2010;50:259-293.
DeVos SL, Goncharoff DK, Chen G, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci 2013;33:12887-12897.
Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron 2012;74:1031-1044.
Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest 2006;116:2290-2296.
Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994;264:1772-1775.
Ding H, Schwarz DS, Keene A, et al. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell 2003;2:209-217.
Miller TM, Kaspar BK, Kops GJ, et al. Virus-delivered small RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann Neurol 2005;57:773-776.
Ralph GS, Radcliffe PA, Day DM, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med 2005;11:429-433.
Raoul C, Abbas-Terki T, Bensadoun JC, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med 2005;11:423-428.
Foust KD, Salazar DL, Likhite S, et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther 2013;21:2148-2159.
Wang H, Yang B, Qiu L, et al. Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum Mol Genet 2014;23:668-681.
Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 2013;12:435-442.
Winer L, Srinivasan D, Chun S, et al. SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol 2013;70:201-207.
Haidet-Phillips AM, Hester ME, Miranda CJ, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 2011;29:824-828.
Gruzman A, Wood WL, Alpert E, et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2007;104:12524-12529.
Bosco DA, Morfini G, Karabacak NM, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 2010;13:1396-1403.
Guareschi S, Cova E, Cereda C, et al. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci U S A 2012;109:5074-5079.
Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci 2013;7:253.
Pickles S, Destroismaisons L, Peyrard SL, et al. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum Mol Genet 2013;22:3947-3959.
Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013;80:415-428.
Riboldi G, Zanetta C, Ranieri M, et al. Antisense oligonucleotide therapy for the treatment of C9ORF72 ALS/FTD diseases. Mol Neurobiol 2014;50:721-732.
Sareen D, O'Rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 2013;5:208ra149.
Raoul C, Estévez AG, Nishimune H, et al. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron 2002;35:1067-1083.
Koval ED, Shaner C, Zhang P, et al. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet 2013;22:4127-4135.
Butovsky O, Jedrychowski MP, Cialic R, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol 2014;77:75-99.
Southwell AL, Skotte NH, Bennett CF, Hayden MR. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med 2012;18:634-643.
Cudkowicz ME, Warren L, Francis JW, et al. Intrathecal administration of recombinant human superoxide dismutase 1 in amyotrophic lateral sclerosis: a preliminary safety and pharmacokinetic study. Neurology 1997;49:213-222.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Reddy, L.V., Miller, T.M. RNA-targeted Therapeutics for ALS. Neurotherapeutics 12, 424–427 (2015). https://doi.org/10.1007/s13311-015-0344-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-015-0344-z