
Abstract
Regeneration of myelin sheaths (remyelination) after central nervous system demyelination is important to restore saltatory conduction and to prevent axonal loss. In multiple sclerosis, the insufficiency of remyelination leads to the irreversible degeneration of axons and correlated clinical decline. Therefore, a regenerative strategy to encourage remyelination may protect axons and improve symptoms in multiple sclerosis. We highlight recent studies on factors that influence endogenous remyelination and potential promising pharmacological targets that may be considered for enhancing central nervous system remyelination.
Similar content being viewed by others
Introduction: The Need for Regenerative Therapy in Multiple Sclerosis
Multiple sclerosis (MS) is a disease of the central nervous system (CNS) characterized by acute focal demyelination, variable remyelination, and extensive neuronal and axonal degeneration [1]. MS affects approximately 2.5 million people worldwide and is thought to be the most commonly acquired neurological disease of young adults.
There are 2 principal components in treating MS: 1) the prevention of damage, usually involving an immunomodulatory approach; and 2) the repair of damage, involving the regeneration of new myelin sheaths (remyelination) [2, 3]. Protection of neurons and their axons, the loss of which is the principal anatomical correlate of progressive clinical deterioration, might be considered a third component [4]. Indeed, the overarching goal of all MS therapy is to identify strategies to prevent axonal loss. Axon protection can be achieved directly as a result of intervention in the mechanisms by which axons are injured or degenerate [5]. However, axon protection can also be achieved as a consequence of immunomodulatory therapies and by the promotion of remyelination.
MS research for many decades has resulted in a considerable investment into the immunological aspects of the disease, as reflected in the identification of several immune-related genes as genetic determinants of disease susceptibility [6] and an understanding of the disease pathogenesis [1, 7]. Although this research has proven to be highly informative and has translated into the development of highly effective immunomodulatory therapies, our knowledge pertaining to aspects of the disease is not directly related to immunology, but it is related to neurobiology instead, and specifically the interdependency between axon and oligodendrocytes remain relatively to be less explored [8, 9]. Thus, although immunomodulatory therapies are proving to be increasingly effective in controlling the initial relapsing-remitting phase of MS, the secondary progressive phase, in which there is continual atrophy of demyelinated axons, remains largely untreatable. Indeed, axon degeneration occurs despite immunomodulatory therapies, suggesting that axon integrity and protection may occur through mechanisms independent of inflammation [10, 11]. Several lines of evidence suggest that the basis of axon atrophy in chronically demyelinated lesions is due, in part, to the absence of myelin-associated trophic signals that are critical for maintaining axon integrity. For example, oligodendrocyte-specific deletions in myelin associated genes PLP, MAG, and CNPase do not cause any obvious defect in myelination, but eventually lead to axonal pathology [12–14]. Moreover, axon degeneration has recently been observed as a consequence of genetically-induced oligodendrocyte-specific ablation [15], thus providing compelling evidence that axon survival is dependent on intact oligodendrocytes, and that axon degeneration in chronically demyelinated lesions can occur independently of inflammation. The presence of low-grade inflammation (mainly cells of the innate immune response) has led to the suggestion that this inflammatory response may also contribute to progressive attritional axonal loss. However, it is far from clear whether this inflammation is a primary cause of axonal degeneration or a secondary response to axonal degeneration occurring due to loss of myelin-associated trophic support. Importantly, the increasing evidence of a role for myelin in preserving axon integrity reveals an evermore pressing need to understand the mechanisms of CNS remyelination.
In addition to preserving axon integrity [16, 17], remyelination also restores saltatory conduction and reverses functional deficits [18–20]. Compelling evidence in support of functional restoration by remyelination has recently been provided by an unusual demyelinating condition in cats in which the reversal of clinical signs is associated with spontaneous remyelination [21]. Taken together these observations imply that an effective means of restoring function and preventing axonal loss in MS is to promote remyelination.
Remyelination as Regenerative Therapy
How might remyelination in MS be enhanced? One proposed approach is to transplant myelinogenic cells of which several types have been described. However, this approach presents a variety of obstacles, including how to achieve: 1) the proper delivery and distribution of cells within a multifocal disease, 2) efficient remyelination by exogenous cells in environments that do not support remyelination by endogenous cells, and 3) a generation of a large numbers of cells that may require immunosuppressive protocols. For these and other reasons, cell transplantation with the primary objective of remyelination is a regenerative strategy for MS fraught with translational difficulties. An alternative approach is to identify ways to enhance remyelination by endogenous myelinating cells. Such an approach requires an understanding of precisely how remyelination is achieved and why it fails.
The principal source of new remyelinating cells is an abundant and widely distributed population of cells in the adult CNS traditionally called oligodendrocyte precursor cells (OPCs). In the adult CNS, these cells are both self-renewing and multipotent, having been observed to give rise to certain neurons, astrocytes (albeit rarely), and Schwann cells, as well as oligodendrocytes in vivo, and so can reasonably be regarded as a type of adult neural stem cell [22–25]. The response to demyelination causes OPCs to become activated, a morphological change accompanied by upregulation of genes not normally expressed in the resting state [26–29]. Activated OPCs proliferate, migrate and rapidly fill up the demyelinated lesions at a density that far exceeds that in normal tissue. To complete the remyelination process, the cells exit the cell cycle and differentiate into myelin sheath-forming oligodendrocytes, which is a complex process involving axon engagement, ensheathment and formation of compacted myelin [30, 31]. As with all regenerative processes the efficiency of remyelination decreases with age primarily due to a decrease in the ability of recruited cells to differentiate [32–35]. This age-associated decline in remyelination resembles and may be in part a determinant of a now well-recognized feature of many chronically demyelinated MS lesions, which are replete with oligodendrocyte lineage cells that are unable to differentiate into myelinating oligodendrocytes [36–39].
Thus, both experimental and clinical pathological studies point to the impairment of precursor differentiation as a common, although not exclusive, feature of insufficient or failed remyelination in MS. This means that identifying pathways involved in the regulation of OPC differentiation represents a critical task in understanding the mechanisms of remyelination and the development of new pharmacological therapies for enhancing endogenous remyelination, and thus axonal protection in MS.
Barriers Preventing Efficient Remyelination
Myelin Debris
The process of primary demyelination creates vast amounts of myelin debris from the unraveling and breakdown of compacted myelin from axons. Myelin fragments in lesions can prevent efficient remyelination by inhibiting oligodendrocyte differentiation. Several lines of evidence reveal that the phagocytic removal of myelin debris is critical for efficient remyelination. First, rapid remyelination that occurs in young animals is associated with efficient removal of myelin debris in contrast to old animals that display slow remyelination and impaired clearance of myelin debris [40, 41]. It is thought that inefficient debris clearance is due to delayed activation and recruitment of phagocytic macrophages. Second, in tissue culture, OPCs plated onto a CNS myelin substrate display significantly impaired differentiation, although their migratory and proliferative properties are unaltered [42]. Third, injection of myelin debris into experimentally induced areas of demyelination in young animals results in impaired remyelination caused by the inhibition of precursor differentiation [43]. Recently, the Fyn-Rho-ROCK and protein kinase C (PKC) signaling pathways has been identified as critical mediators of myelin-associated inhibition [44].
Hyaluronan
The glycosaminoglycan hyaluronan is another component found to inhibit OPC differentiation and remyelination [45]. Hyaluronan has been observed to accumulate in chronic MS lesions and in mice with experimental autoimmune encephalomyelitis (EAE). High molecular weight hyaluronan can directly inhibit OPC differentiation in culture. A recent study has found that hyaluronan inhibits OPC differentiation by binding to toll-like receptor 2 (TLR2) in oligodendrocyte lineage cells in culture and that hyaluronidases produced by OPCs convert high molecular weight hyaluronan to the more inhibitory low molecular weight form [46]. Moreover, inhibition of TLR2 and its signaling pathways can block hyaluronan inhibition to enable OPC differentiation. In MS lesions, TLR2 is upregulated and expressed by oligodendrocytes, thus suggesting that hyaluronan-mediated inhibition of OPC differentiation may occur through TLR2 signaling.
Semaphorins
Semaphorins have been implicated as key regulators of several stages of remyelination. Both experimental functional studies and expression analysis of MS lesions support a model in which recruitment is regulated by an appropriate balance of semaphorin 3A, a repulsive guidance cue, which if it becomes the dominant form may prevent precursors entering areas of demyelination, and semaphorin 3F, an attractive guidance cue that facilitates recruitment [47, 48]. Semaphorin 3A has also been shown to be a potent inhibitor of precursor differentiation (a role which paradoxically might be advantageous to recruitment by preventing cells leaving the cell cycle) and therefore a potential therapeutic target for overcoming remyelination block in nonremyelinating MS lesions [49].
Although the concept of these or indeed other inhibitors of differentiation, such as poly-sialated neural cell adhesion molecule (PSA-NCAM) [50], as causes of remyelination failure are derived mainly from experimental studies and inferred from descriptive observations of MS tissues. Nevertheless, these studies raise important questions as to the existence of signaling pathways in OPCs that respond to extrinsic inhibitory factors, thus offering possible therapeutic targets to overcome remyelination block in the hostile environment of MS lesions.
Pharmacological Targets of Remyelination
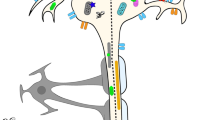
Several regulators of CNS remyelination have been recently identified that potentially lend themselves to pharmacological manipulation, including LINGO-1, Notch1, Wnt, and retinoid X receptors (RXRs). These signaling pathways are particularly interesting because pharmacological agonists or antagonists against are available (or are currently being developed) for treatment against other diseases, such as cancer. LINGO-1, Notch, and Wnt signaling in OPCs are negative regulators of OPC differentiation, whereas RXRs are positive regulators of differentiation. By targeting these pathways, it might be possible to pharmacologically encourage remyelination from endogenous OPCs as a regenerative strategy to treat MS (Fig. 1).

Pharmacological targets of central nervous system (CNS) remyelination. The diagram illustrates the recruitment of an oligodendrocyte precursor cell (OPC) to an area of demyelination, and its differentiation into a remyelinating oligodendrocyte. Chronically demyelinated axons that do not acquire remyelinated sheaths are liable to undergo degeneration. Conversely, stimulation of remyelination preserves the integrity of axons. Several molecular pathways known to regulate oligodendrocyte differentiation are possible pharmaceutical targets of CNS remyelination. Semaphorin 3A and 3F are involved in OPC recruitment into the lesion. Lingo-1, Wnt/β-catenin, Semaphorin 3A, and canonical Notch signalling pathways negatively regulate CNS remyelination by preventing OPC differentiation, whereas retinoid X receptor (RXR)-γ and the noncanonical Notch signalling pathways enhance CNS remyelination by stimulating OPC differentiation
LINGO-1
LINGO-1 was identified in a search for CNS-specific leucine rich repeat proteins [51]. It has been shown to regulate axon outgrowth by interaction with the Nogo-66 receptor complex. More recently LINGO-1 has also been found to be a negative regulator of oligodendrocyte differentiation [52]. Treatment of cultured OPCs with small interference RNAs (siRNAs) generated against LINGO-1, dominant negative LINGO-1, or soluble LINGO (LINGO-Fc) resulted in increased morphological differentiation of oligodendrocytes as characterized by the abundance of terminal membrane sheets. Mice deficient in LINGO-1 or treated with an antibody antagonist against LINGO-1 exhibited increased remyelination and functional recovery from EAE, a model of immune-mediated demyelination [53]. Moreover, the LINGO-1 antagonist is able to promote CNS remyelination by directly stimulating OPC differentiation in nonimmune, toxin-induced models of demyelination in rats [54]. These findings reveal the importance of LINGO-1 signaling in regulating OPC differentiation in the injured CNS. Moreover, the restricted expression of LINGO-1 in the CNS makes therapeutic targeting of LINGO-1 potentially advantageous for improving myelin repair without affecting non-neural tissues.
Wnt Signaling
A screen for transcription factors associated with CNS remyelination in experimentally demyelinated mice led to the identification of Tcf4 expression in oligodendrocyte lineage cells and the elucidation of the canonical Wnt pathway as a negative regulator of precursor differentiation [55]. Tcf4 is expressed in the mouse white matter immediately after birth, but it is barely detectable in the adult [55–57]. However, following CNS injury, Tcf4 is re-expressed and upregulated in OPCs that are recruited to the lesion [56]. Moreover, Tcf4 is highly expressed in active MS lesions, suggesting a role in remyelination. In response to Wnt activation, Tcf4 is thought to interact with β-catenin in the nucleus to regulate transcription of Wnt-β-catenin pathway target genes [58]. Indeed, disruption of Wnt signaling in oligodendrocyte lineage cells by using transgenic mice that actively express a dominant active β-catenin gene results in mice displaying severe tremor and ataxia within 1 week after birth due to delayed oligodendrocyte differentiation and hypomyelination [55]. However, this effect is transient as CNS myelination ultimately catches up and appears normal in adult mice. Experimental demyelination performed on these transgenic mice results in a similar delay in oligodendrocyte differentiation and remyelination, without affecting OPC recruitment to lesions. These findings have opened up a new area of research to dissect the mechanism of Wnt signaling in OPC differentiation. But more importantly, given the growing interest to develop pharmacological inhibitors against the Wnt pathway in cancer therapy [59], it might be possible in the near future to use Wnt inhibitors to stimulate OPC differentiation and improve CNS remyelination in MS.
Notch Signaling
One of the potential contributors of remyelination efficiency in MS is thought to be reactive astrocytosis in lesions, which can potentially have both beneficial and detrimental roles [60, 61]. In search of putative astrocytic inhibitors of remyelination, a microarray analysis was performed on purified human astrocytes treated with cytokines that are known to be significantly upregulated in the brains of MS patients [62]. This led to the identification of the Notch ligand, Jagged in astrocytes, which is upregulated in response to the cytokine TGF-β. Notch1 and its effector Hes5 were detected in immature oligodendrocytes of MS lesions and following demyelination in mice [62, 63]. When cultured human OPCs were exposed to Jagged, these OPCs failed to mature [62]. However, the role of Notch1 signaling on OPC differentiation during remyelination in vivo has remained inconclusive for several reasons. First, remyelination can occur with great efficiency despite the expression of Notch in precursor cells and Jagged expression within the demyelinated environment [63, 64]. Second, conditional ablation of Notch in precursors leads to no [63] or a relatively minor effect on remyelination efficiency [65]. Third, when mice with experimental autoimmune encephalomyelitis (a model of immune mediated demyelination) were treated with a γ-secretase inhibitor, MW167, which prevents Notch signaling, myelin repair and axonal survival were improved [66]. However, Notch is also expressed by cells associated with inflammation [67], thus it remains possible, even likely that the mechanism by which MW167 enhances CNS myelin repair occurs through immunomodulation rather than by directly stimulating OPC differentiation. It would be of interest to assess the effect of MW167 directly on OPC differentiation and myelination by using purified OPC cultures or myelinating co-cultures. Fourth, noncanonical Notch signaling in OPCs is involved in the induction of OPC differentiation [68]. Indeed, evidence from careful examination of MS tissue implies a failure of Notch signaling as an explanation for incomplete differentiation [69]. Within OPCs present in nonrepairing lesions, the Notch intracellular domain accumulates outside the nucleus and therefore cannot activate the expression of downstream targets. The perinuclear aggregation of the Notch intracellular domain is attributed to an increased expression of TAT-interacting protein 30 kDa (TIP30), which leads to an inhibition of the nuclear transporter importin-β. Thus, the value of Notch signaling as a therapeutic target for remyelination remains uncertain
Retinoid X Receptors (RXRs)
Thus far, most studies have focused on the negative regulation of OPC differentiation in remyelination. Recently, RXRs have been demonstrated as positive regulators of OPC differentiation. RXRs are nuclear receptors that regulate cell proliferation and differentiation [70, 71]. There are 3 members in the RXR family (RXRα, RXRβ, and RXRγ) that form homodimers or heterodimers with other nuclear receptors, including retinoic acid receptors (RAR), thyroid hormone receptors, vitamin D receptors (VDR), peroxisome proliferator activator proteins (PPAR), and liver X receptors (LXR), to control transcription of target genes. Following CNS injury, all 3 members of RXR are highly expressed in lesions [72]. We have recently found that RXRγ is highly expressed in oligodendrocyte lineage cells during the regenerative phase of CNS remyelination by using microarray analysis of the separate stages of CNS remyelination in rats. In acute and remyelinating MS lesions, RXRγ is highly expressed by oligodendrocyte lineage cells, macrophages, and astrocytes, but its expression is very low in chronic inactive lesions, suggesting RXRγ is an active component of CNS repair. Transfection of cultured OPCs with small interference RNAs generated against RXRγ resulted in less morphologically differentiated oligodendrocytes. Analysis of RXRγ knockout mice that have received focal CNS demyelination resulted in the accumulation of undifferentiated OPCs and less mature oligodendrocytes in lesions. These results indicate RXRγ regulates oligodendrocyte differentiation.
The mechanisms of RXR signaling and its regulation during CNS remyelination are presently unclear. As with other nuclear receptors, RXRs are ligand-induced transcription factors that function through heterodimeric association with other nuclear receptors. However, RXR is seated at the center of nuclear receptor activity, because it can heterodimerize with a wide range of receptors, including members of RAR, thyroid hormone receptor, PPAR, LXR, and VDR. Once activated, the RXR heterodimeric complex is able to induce transcription of genes associated with cell differentiation. Moreover, RXRs are able to function as either permissive or nonpermissive heterodimers. In permissive dimerization, such as RXR:PPAR or RXR:LXR, the complex can be activated by RXR selective ligands, such as 9cRA, whereas in nonpermissive dimerization, such as RXR:RAR, RXR:thyroid hormone receptors or RXR:VDR, the complex can only be activated by ligands selective for the partners of RXR, and not by RXR selective ligands alone, so that RXR activity is subordinated under nonpermissive dimerization. Because we found that RXR activation enhances CNS remyelination, the mechanism by which RXR signaling occurs in oligodendrocytes is likely through permissive heterodimerization, such binding to PPAR or LXR. Interestingly, PPARγ, which is also expressed by oligodendrocyte lineage cells, has been shown to accelerate oligodendrocyte differentiation when it is activated [73]. Moreover, deletion of LXRγ and LXRβ in mice results in dysmyelination or myelin abnormalities [74]. Identification of the RXR heterodimeric partner would therefore improve our understanding of RXR signaling in oligodendrocyte lineage cells, and help develop more finely tuned agonists that would target oligodendrocytes specifically in remyelination therapy.
Chemical agonists and antagonists of RXR signaling, or rexinoids, are widely available and are showing promise in the treatment of certain types of cancers and metabolic disorders [75]. When cultured OPCs are exposed to the RXR selective antagonists (HX531 and PA452), oligodendrocyte differentiation is severely impaired. By contrast, when OPCs are exposed to the RXR agonists, 9-cis-retinoic acid (9cRA), HX630, or PA024, oligodendrocytes are stimulated to differentiate and form myelin membrane-like sheets in culture. Further experiments testing the effect of 9cRA on ex vivo cerebellar slice cultures of demyelination, as well as on rats that have received focal demyelination in the CNS, resulted in the significant increase of remyelinated axons. Moreover, axons in lesions displayed thicker myelin sheaths in 9cRA treated rats compared to controls at an early stage of remyelination, suggesting 9cRA accelerated CNS remyelination. It is known that RXR agonists can also attenuate inflammation by regulating macrophage activity [76]. Immunomodulation and acceleration of OPC differentiation are the crucial ingredients needed for successful MS treatment. Rexinoids may exert dual functions in regulating inflammation and OPC differentiation in the injured CNS, thus showing promise as feasible regenerative therapeutics to improve CNS remyelination in demyelinating diseases such as MS.
Conclusions and Future Prospects
It is now widely acknowledged that inducing differentiation of oligodendrocyte lineage cells present within areas of demyelination into remyelinating oligodendrocytes represents a major focus of therapeutic remyelination research. Potentially this could be achieved either by overcoming putative inhibitors of differentiation present within lesions or by the administration of agents that induce differentiation. Critical to achieving this objective is the identification of pathways by which oligodendrocyte differentiation is controlled. In this review, we have identified several pathways associated with preventing differentiation for which the formulation of effective antagonists would be expected to have therapeutic efficacy. However, much work is yet to be done to achieve a comprehensive understanding of molecular logic underlying optimal remyelination. In this regard, genomic [77], gene expression [78] and proteomic [79] analyses of MS patients will be critical.
Developments in the pharmacology of “patient-tailored” cancer treatment may provide such regenerative medicines in a shorter time frame than might have been predicted several years ago. Differentiation therapies have been attracting considerable interest in cancer medicine and are predicated on the hypothesis that inducing a cancer cell to differentiate will either convert it into a nondividing cell or, more likely, cause it to undergo apoptosis [80]. The objectives of cancer differentiation therapies in promoting cell differentiation are thus strikingly similar to those of regenerative medicine and especially the enhancement of remyelination. One can therefore envisage a situation in which drugs developed for cancer therapy can be re-designated for remyelination therapy. As a specific example, several cancers, including the majority of colon adenocarcinomas, arise due to gain-of-function mutations in the canonical Wnt signaling pathway. Pharmacological inhibitors of Wnt pathway are being actively developed in cancer medicine and it is possible that these may also find a role in enhancing remyelination by inhibiting Wnt signaling in CNS precursors and normalizing the kinetics of myelin regeneration [81, 82].
Currently RXR agonists are being tested for the treatment of certain cancers, and are also thought to be useful in the treatment of metabolic disorders [75]. A licensed RXR agonist Targretin (bexarotene, Eisai Inc., Japan) is already in clinical use for the treatment of cutaneous T-cell lymphoma [83]. In EAE, a well-established model of immune-mediated demyelination that mimics many aspects of MS, 9cRA has been shown to suppress inflammation. Our work suggests RXR agonists will be of value as a regenerative therapy, filling the unoccupied niche of a treatment for progressive MS [84]. Thus, Targretin and other RXR agonists could conceivably provide an important advance in MS therapy, contributing not only to damage suppression through immunomodulation, but also axon preservation through enhancement of remyelination.
In summary, we have focused on those pathways that have been shown to regulate the regenerative process of remyelination. There are an increasing number of pathways and mechanisms being identified that could potentially play similar roles [85–88]. As previously indicated, such research could impact not only MS, but other myelin disorders, such as periventricular leukomalacia (PVL), in which a critical window for repair is inadequately addressed by current therapies. Therefore, the next few years promise to be an exciting time for remyelination research with the promise of developing a much better understanding of the pathobiology of failed remyelination, as well as development of new pharmacological treatments by which this important regenerative process can be promoted.
References
Compston A, Coles A. Multiple sclerosis. Lancet 2008;372:1502–1517.
Martino G, Franklin RJM, Baron van Evercooren A, Kerr D, Group SC. Stem cell transplantation in multiple sclerosis: current status and future prospects. Nat Rev Neurol 2010;6:247–255.
Franklin RJM, ffrench-Constant C. Stem cell treatments and multiple sclerosis. BMJ 2010;340:986–985.
Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 2008;31:247–269.
Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 2006;7:932–941.
Oksenberg JR, Baranzini SE, Sawcer S, Hauser SL. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat Rev Genet 2008;9:516–526.
Coles AJ, Compston DA, Selmaj KW, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med 2008;359:1786–1801.
Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci 2008;31:535–561.
Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci 2010;11:275–283.
Coles AJ, Wing MG, Molyneux P, et al. Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol 1999;46:296–304.
Dutta R, Trapp BD. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology 2007;68(22 suppl 3):S22-S31.
Lappe-Siefke C, Goebbels S, Gravel M, et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 2003;33:366–374.
Griffiths I, Klugmann M, Anderson T, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 1998;280:1610–1613.
Li C, Tropak MB, Gerlal R, et al. Myelination in the absense of myelin-associated glycoprotein. Nature 1994;369:747–750.
Pohl HB, Porcheri C, Mueggler T, et al. Genetically induced adult oligodendrocyte cell death is associated with poor myelin clearance, reduced remyelination, and axonal damage. J Neurosci 2011;31:1069–1080.
Edgar JM, Nave KA. The role of CNS glia in preserving axon function. Curr Opin Neurobiol 2009;5:498–504.
Irvine KA, Blakemore WF. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008;131:1464–1477.
Liebetanz D, Merkler D. Effects of commissural de- and remyelination on motor skill behaviour in the cuprizone mouse model of multiple sclerosis. Exp Neurol 2006;202:217–224.
Jeffery ND, Crang AJ, O'Leary MT, Hodge SJ, Blakemore WF. Behavioural consequences of oligodendrocyte progenitor cell transplantation into demyelinating lesions in rat spinal cord. Eur J Neurosci 1999;11:1508–1514.
Smith KJ, Blakemore WF, McDonald WI. Central remyelination restores secure conduction. Nature 1979;280:395–396.
Duncan ID, Brower A, Kondo Y, Curlee JF Jr, Schultz RD. Extensive remyelination of the CNS leads to functional recovery. Proc Natl Acad Sci U S A 2009;106:6832–6836.
Rivers LE, Young KM, Rizzi M, et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci 2008;11:1392–1401.
Nunes MC, Roy NS, Keyoung HM, et al. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med 2003;9:439–247.
Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 2008;135:145–157.
Tripathi RB, Rivers LE, Young KM, Jamen F, Richardson WD. NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J Neurosci 2010;30:16383–16390.
Glezer I, Lapointe A, Rivest S. Innate immunity triggers oligodendrocyte progenitor reactivity and confines damages to brain injuries. FASEB J 2006;20:750–752.
Watanabe M, Hadzic T, Nishiyama A (2004) Transient upregulation of Nkx2.2 expression in oligodendrocyte lineage cells during remyelination. Glia 46:311–322.
Zhao C, Fancy SPJ, Franklin RJM, ffrench-Constant C. Upregulation of oligodendrocyte precursor cell αv integrin and its extracellular ligands during CNS remyelination. J Neurosci Res 2009;87:3447–3455.
Fancy SPJ, Zhao C, Franklin RJM. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol Cell Neurosci 2004;27:247–254.
Crockett DP, Burshteyn M, Garcia C, Muggironi M, Casaccia-Bonnefil P. Number of oligodendrocyte progenitors recruited to the lesioned spinal cord is modulated by the levels of the cell cycle regulatory protein p27Kip-1. Glia 2005;49:301–308.
Chen Y, Wu H, Wang S, et al. The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat Neurosci 2009;12:1398–406.
Woodruff RH, Fruttiger M, Richardson WD, Franklin RJM. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci 2004;25:252–262.
Sim FJ, Zhao C, Penderis J, Franklin RJM. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci 2002;22:2451–2459.
Shields SA, Gilson JM, Blakemore WF, Franklin RJM. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia 1999;28:77–83.
Shen S, Sandoval J, Swiss V, et al. Age-dependent epigenetic control of differentiation inhibitors: a critical determinant of remyelination efficiency. Nat Neurosci 2008;11:1024–1034.
Kuhlmann T, Miron V, Cuo Q, Wegner C, Antel J, Bruck W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008;131:1749–58.
Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 2002;346:165–173.
Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci 1998;18:601–609.
Scolding N, Franklin R, Stevens S, Heldin CH, Compston A, Newcombe J. Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain 1998;121:2221–2228.
Gilson J, Blakemore WF. Failure of remyelination in areas of demyelination produced in the spinal cord of old rats. Neuropathol Appl Neurobiol 1993;19:173–181.
Zhao C, Li WW, Franklin RJM. Differences in the early inflammatory responses to toxin-induced demyelination are associated with the age-related decline in CNS remyelination. Neurobiol Aging 2006;27;1298–1307.
Robinson S, Miller RH. Contact with central nervous system myelin inhibits oligodendrocyte progenitor maturation. Dev Biol 1999;216:359–368.
Kotter MR, Li WW, Zhao C, Franklin RJM. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci 2006;26:328–232.
Baer AS, Syed YA, Kang SU, et al. Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling. Brain 2009;132:465–481.
Back SA, Tuohy TM, Chen H, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med 2005;11:966–972.
Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A 2010;107:11555–11560.
Williams A, Piaton G, Aigrot MS, et al. Semaphorin 3A and 3F: key players in myelin repair in multiple sclerosis? Brain 2007;130:2554–2565.
Piaton G, Aigrot MS, Williams A, et al. Class 3 semaphorins influence oligodendrocyte precursor recruitment and remyelination in adult central nervous system. Brain 2011;134:1156–1167.
Syed YA, Hand E, Mobius W, et al. Inhibition of CNS Remyelination by the Presence of Semaphorin 3A. J Neurosci 2011;31:3719–3728.
Charles P, Reynolds R, Seilhean D, et al. Re-expression of PSA-NCAM by demyelinated axons: an inhibitor or remyelination in multiple sclerosis? Brain 2002;125:1972–1979.
Mi S, Lee X, Shao Z, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 2004;7:221–228.
Mi S, Miller RH, Lee X, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci 2005;8:745–751.
Mi S, Hu B, Hahm K, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med 2007;13:1228–1233.
Mi S, Miller RH, Tang W, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol 2009;65:304–315.
Fancy SPJ, Baranzini SE, Zhao C, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev 2009;23:1571–1585.
Ye F, Chen Y, Hoang T, et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci 2009;12:829–838.
Fu H, Cai J, Clevers H, et al. A genome-wide screen for spatially restricted expression patterns identifies transcription factors that regulate glial development. J Neurosci 2009;29:11399–11408.
Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004;303:1483–1487.
Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov 2006;5:997–1014.
Franklin RJM, Crang AJ, Blakemore WF. The role of astrocytes in the remyelination of glia-free areas of demyelination. Adv Neurol 1993;59:125–33.
Williams A, Piaton G, Lubetzki C. Astrocytes — friends or foes in multiple sclerosis? Glia 2007;55:1300–1312.
John GR, Shankar SL, Shafit-Zagardo B, et al. Multiple sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med 2002;8:1115–1121.
Stidworthy MF, Genoud S, Li WW, et al. Notch1 and Jagged1 are expressed after CNS demyelination but are not a major rate-determining factor during remyelination. Brain 2004;127;1928–1941.
Seifert T, Bauer J, Weissert R, Fazekas F, Storch MK (2006) Notch1 and its ligand Jagged1 are present in remyelination in a T-cell- and antibody-mediated model of inflammatory demyelination. Acta Neuropathol(Berl) 113:195–203.
Zhang Y, Argaw AT, Gurfein BT, et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc Natl Acad Sci U S A 2009;106:19162–19167.
Jurynczyk M, Jurewicz A, Bielecki B, Raine CS, Selmaj K. Inhibition of Notch signaling enhances tissue repair in an animal model of multiple sclerosis. J Neuroimmunol 2005;170:3–10.
Hoyne GF, Dallman MJ, Champion BR, Lamb JR. Notch signalling in the regulation of peripheral immunity. Immunol Rev 2001;182:215–227.
Hu QD, Ang BT, Karsak M, et al. F3/Contactin acts as a functional ligand for notch during oligodendrocyte maturation. Cell 2003;115:163–175.
Nakahara J, Kanekura K, Nawa M, Aiso S, Suzuki N. Abnormal expression of TIP30 and arrested nucleocytoplasmic transport within oligodendrocyte precursor cells in multiple sclerosis. J Clin Invest 2009;119:169–181.
Germain P, Chambon P, Eichele G, et al. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol Rev 2006;58:760–772.
Lefebvre P, Benomar Y, Staels B. Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol Metab 2010;21:676–683.
Schrage K, Koopmans G, Joosten EA, Mey J. Macrophages and neurons are targets of retinoic acid signaling after spinal cord contusion injury. Eur J Neurosci 2006;23:285–295.
Saluja I, Granneman JG, Skoff RP. PPAR delta agonists stimulate oligodendrocyte differentiation in tissue culture. Glia 2001;33:191–204.
Wang L, Schuster GU, Hultenby K, Zhang Q, Andersson S, Gustafsson JA. Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc Natl Acad Sci U S A 2002;99:13878–13883.
Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov 2007;6:793–810.
Xu J, Drew PD. 9-Cis-retinoic acid suppresses inflammatory responses of microglia and astrocytes. J Neuroimmunol 2006;171:135–144.
Baranzini SE, Wang J, Gibson RA, et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet 2009;18:767–778.
Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 2002;8:500–508.
Han MH, Hwang SI, Roy DB, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 2008;451:1076–1081.
van Es JH, Clevers H. Notch and Wnt inhibitors as potential new drugs for intestinal neoplastic disease. Trends Mol Med 2005;11:496–502.
Takahashi-Yanaga F, Sasaguri T. The Wnt/beta-catenin signaling pathway as a target in drug discovery. J Pharmacol Sci 2007;104:293–302.
Huang SM, Mishina YM, Liu S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009;461:614–620.
Ballanger F, Nguyen JM, Khammari A, Dreno B. Evolution of clinical and molecular responses to bexarotene treatment in cutaneous T-cell lymphoma. Dermatology 2010;220:370–375.
Diab A, Hussain RZ, Lovett-Racke AE, Chavis JA, Drew PD, Racke MK. Ligands for the peroxisome proliferator-activated receptor-gamma and the retinoid X receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. J Neuroimmunol 2004;148:116–126.
Lecca D, Trincavelli ML, Gelosa P, et al. The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS One 2008;3:e3579.
Pasquini LA, Paez PM, Moreno MA, Pasquini JM, Soto EF. Inhibition of the proteasome by lactacystin enhances oligodendroglial cell differentiation. J Neurosci 2003;23:4635–4644.
Gadea A, Aguirre A, Haydar TF, Gallo V. Endothelin-1 regulates oligodendrocyte development. J Neurosci 2009;29:10047–10062.
Sabo JK, Aumann TD, Merlo D, Kilpatrick TJ, Cate HS. Remyelination is altered by bone morphogenic protein signaling in demyelinated lesions. J Neurosci 2011;31:4504–4510.
Acknowledgement
Full conflict of interest disclosure is available in the electronic supplementary material for this article.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
ESM 1
(PDF 510 kb)
Rights and permissions
About this article
Cite this article
Huang, J.K., Fancy, S.P.J., Zhao, C. et al. Myelin Regeneration in Multiple Sclerosis: Targeting Endogenous Stem Cells. Neurotherapeutics 8, 650–658 (2011). https://doi.org/10.1007/s13311-011-0065-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-011-0065-x