
Abstract
Introduction
Real-world data provide insight into how medications perform in clinical practice. The PIONEER REAL Switzerland study aimed to understand clinical outcomes with oral semaglutide in adults with type 2 diabetes (T2D).
Methods
PIONEER REAL Switzerland was a 34–44-week, multicentre, prospective, non-interventional, single-arm study of adults with T2D naïve to injectable glucose-lowering medication who were initiated on oral semaglutide in routine clinical practice. The primary endpoint was change in glycated haemoglobin (HbA1c) from baseline (BL) to end of study (EOS); secondary endpoints included change in body weight (BW) from BL to EOS and the proportion of participants achieving HbA1c < 7.0% and the composite endpoints HbA1c reduction ≥ 1%-points with BW reduction ≥ 3% or ≥ 5% at EOS. Safety was assessed in participants who received ≥ 1 dose of oral semaglutide.
Results
Of the 185 participants (female/male, n = 67/118) initiating oral semaglutide, 168 (90.8%) completed the study and 143 (77.3%) remained on treatment with oral semaglutide at EOS. At BL, participants had a mean age of 62 years, diabetes duration of 6.4 years, HbA1c of 7.7%, BW of 95.6 kg and body mass index of 33.2 kg/m2; 56.2% of participants were receiving glucose-lowering medications. Significant reductions were observed for HbA1c (estimated change − 0.91%; 95% confidence interval [CI] − 1.10, − 0.71; p < 0.0001) and BW (estimated change − 4.85%; 95% CI − 5.70, − 4.00; p < 0.0001). In total, 139 adverse events (AEs) were reported in 65 (35.1%) participants; most were mild or moderate. The most frequent AEs were gastrointestinal disorders (27.0%); 31 AEs in 20 (10.8%) participants led to discontinuation of oral semaglutide. Six serious AEs were reported; all were considered unlikely to be related to oral semaglutide.
Conclusion
People living with T2D treated with oral semaglutide in Switzerland achieved clinically significant reductions in HbA1c and BW, with no new safety signals.
Clinical Trial Registration
ClinicalTrials.gov: NCT04537624.
A graphical abstract is available for this article.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
In the phase 3 PIONEER programme, oral semaglutide demonstrated efficacy versus placebo and most active comparators in achieving glycaemic control and body weight loss, with a safety profile consistent with the glucagon-like peptide-1 receptor agonist drug class. |
Real-world data on the use of oral semaglutide in clinical practice are lacking. The PIONEER REAL programme is investigating the use of oral semaglutide in the real-world setting in 13 countries worldwide. |
As part of this programme, the non-interventional PIONEER REAL Switzerland study assessed clinical outcomes associated with the once-daily use of oral semaglutide initiated within routine clinical practice in adults with type 2 diabetes in Switzerland. |
What was learned from the study? |
Participants treated with oral semaglutide in routine clinical practice in Switzerland experienced clinically significant improvements in glycaemic control and body weight, with a safety profile consistent with that reported in phase 3 trials. |
The findings of PIONEER REAL Switzerland complement the results of the phase 3 PIONEER programme and support the use of oral semaglutide in a real-world setting. |
Digital Features
This article is published with digital features, including a graphical abstract, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.24807525.
Introduction
In Switzerland, there were an estimated 389,000 adults living with diabetes in 2021, with this predicted to rise to nearly half a million by 2045 [1].
The achievement and maintenance of glycaemic control is the key target for people living with type 2 diabetes (T2D), with weight loss also an important target for people with overweight and obesity [2, 3]. There is compelling evidence that these factors can reduce the risk of long-term complications, including cardiovascular (CV) and microvascular disease [4]. The Swiss recommendations highlight that nearly all individuals with T2D are at high CV risk and therefore advise cardiorenal protection for all people living with T2D [5]. Initial treatment should be combination treatment with metformin and either a sodium-glucose co-transporter-2 inhibitor (SGLT2i) or a glucagon-like peptide-1 receptor agonist (GLP-1 RA) [5]. In addition to improving glycaemic control and weight loss, the pleiotropic effects of GLP-1 RA include reductions in blood pressure, inflammation and postprandial lipaemia, all of which may contribute to a reduction in CV risk [6].
Semaglutide is the first GLP-1 RA available as a once-daily oral (3, 7 and 14 mg as an adjunct to diet and exercise) [7,8,9] formulation for the treatment of T2D. Subcutaneous semaglutide was first approved by Swissmedic, the Swiss regulatory authority, in 2018 as an adjunct to diet and exercise for the treatment of T2D [10, 11], with the oral formulation receiving approval in March 2020 [9, 12]. The approval of oral semaglutide was based on the comprehensive phase 3 PIONEER clinical development programme [9], which demonstrated the efficacy of oral semaglutide versus placebo and most active comparators in achieving glycaemic control and weight loss, with a safety profile consistent with the GLP-1 RA drug class [9, 13,14,15,16,17,18].
PIONEER REAL comprises 13 non-interventional studies designed to support the phase 3 PIONEER clinical programme, each in a different country, and is investigating the use of oral semaglutide in a real-world setting, in a population of adults aged ≥ 18 years living with T2D who have not previously been treated with injectable glucose-lowering medications.
As part of the programme, the non-interventional PIONEER REAL Switzerland study assessed clinical outcomes associated with the use of once-daily oral semaglutide within routine clinical practice in adults with T2D in Switzerland.
Methods
Study Design
PIONEER REAL Switzerland was a 34–44-week, multicentre, prospective, non-interventional, single-arm study across 19 primary and specialist care centres in Switzerland. The study protocol was approved by the appropriate health authorities according to local guidelines and by an Institutional Review Board/Independent Ethics Committee. The study was conducted in accordance with the Declaration of Helsinki and International Council on Harmonisation Good Clinical Practice guidelines. Participants provided written informed consent prior to commencement of any study-related activity. The study is registered with ClinicalTrials.gov (NCT04537624).
Participants
Adults aged ≥ 18 years with a diagnosis of T2D were eligible for inclusion if: they were treatment-naïve to injectable glucose-lowering drug(s); they had an available glycated haemoglobin (HbA1c) value within 90 days prior to or taken at the informed consent and treatment initiation visit (visit 1) in line with local clinical practice; and the decision to initiate treatment with oral semaglutide was made by the treating physician and patient or legally acceptable representative and based on local label before, and independently from, the decision to include the patient in this study. Full eligibility criteria are reported in Electronic Supplementary Material (ESM) Table S1.
Study Procedures
Participants received once-daily oral semaglutide in accordance with local clinical practice, with no additional diagnostic or monitoring procedures. Oral semaglutide was not provided by the sponsor. The decisions to prescribe other glucose-lowering treatments, diet and exercise were at the discretion of the treating physician. The treating physician determined the starting dose, dose escalation and maintenance dose, as well as any subsequent changes to the maintenance dose. Participants attended an informed consent and treatment initiation visit, a number of intermediate visits depending on the local clinical practice and an end of study (EOS) visit. The first visit within the window from weeks 34–44 was considered the EOS visit (ESM Fig. S1). Due to the impact of the coronavirus disease 2019 (COVID-19) pandemic, EOS visits outside the 34–44-week window were allowed. If a HbA1c measurement was unavailable in the 34–44-week window, the first HbA1c measurement taken thereafter and up until the last patient last visit were recorded.
Endpoints and Assessments
The primary endpoint was the change in HbA1c (%-points) from baseline to EOS. Secondary endpoints included relative change in body weight (%) from baseline to EOS; absolute change in body weight (kg) from baseline to EOS; percentage of participants achieving HbA1c < 7% at EOS; and the composite endpoints of (1) participants achieving HbA1c reduction of ≥ 1%-points and body weight reduction of ≥ 5% from baseline to EOS and (2) participants achieving combined HbA1c reduction of ≥ 1%-points and body weight reduction of ≥ 3% or ≥ 5% from baseline to EOS. Exploratory endpoints were treatment with oral semaglutide at EOS (yes/no), oral semaglutide dose (mg/day) at EOS, addition of new glucose-lowering medication or increased baseline glucose-lowering medication dose (other than oral semaglutide) during the study period at EOS (yes/no), cessation or dose reduction of baseline glucose-lowering medication dose during the study period at EOS (yes/no), clinical success as assessed by the physician at EOS (yes/no), change in waist circumference (cm) from baseline to EOS and self-reported severe hypoglycaemia (defined as an episode of hypoglycaemia requiring the assistance of another person to actively administer carbohydrate or glucagon, or take other corrective action) during the study period (yes/no).
Statistical Analysis
The sample size was determined based on a criterion of 90% probability of obtaining a 95% confidence interval (CI) for the change in HbA1c from baseline with a maximum half-width of 0.30. The standard deviation (SD) of the mean change in HbA1c was assumed to be 1.7%-points, with the added assumption of an expected greater variation due to differences in whether participants discontinue treatment or not. Based on these assumptions, the number of participants needed for analysis was 145. Since data were to be collected as part of routine clinical practice, the proportion of participants with an available EOS HbA1c measurement was assumed to be 75% based on unpublished data, meaning that 194 participants were needed to ensure 145 participants with an available HbA1c EOS measurement.
The full analysis set (FAS) included all eligible participants who signed the informed consent and initiated treatment with oral semaglutide. The primary analysis of the primary, secondary and exploratory endpoints was based on the FAS in-study observation period, i.e. the time period when participants were considered in the study regardless of potential discontinuation of oral semaglutide. A secondary analysis was based on the on-treatment observation period, i.e. the time period when participants were considered treated with oral semaglutide. Participants lost to follow-up were considered not to have completed the study. Primary and secondary analyses of the primary endpoint used a mixed model for repeated measures (MMRM) for the FAS. A sensitivity analysis was also performed for the primary endpoint using a pattern-mixture model based on the in-study FAS. Because of the COVID-19 pandemic, an additional sensitivity analysis using a MMRM based on the in-study FAS-EOS visit within the original time window was performed to explore any impact of the COVID-19-related amendment on the primary endpoint. Estimated response and change in response were analysed using baseline HbA1c value, age, baseline body mass index (BMI), time and time-squared as covariates and sex, oral glucose-lowering drugs at baseline, diabetes duration and site as fixed factors with random intercept and time (slope). Continuous secondary and exploratory endpoints were analysed similarly to the primary endpoint but with the corresponding equivalent baseline value instead of the original baseline value; categorical endpoints were reported as frequency tables (percentages with numerator counts).
Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
Participants
Between 24 August 2020 and 11 January 2023, 197 people signed informed consent, of whom 185 met the eligibility criteria and initiated treatment with oral semaglutide. The study was completed by 168 participants (90.8%), with 143 (77.3%) remaining on treatment with oral semaglutide at EOS. A total of 34 participants (18.4%) discontinued oral semaglutide: 22 (11.9%) because of safety concerns, two (1.1%) due to a change in treatment strategy and one (0.5%) due to a change in reimbursement status; for the remaining nine patients (4.8%) who discontinued treatment, the reason for discontinuation was unknown. The total combined follow-up time for the in-study observation period was 5852.0 weeks; the mean (SD) follow-up time was 31.6 (13.53) weeks (median 36 weeks) for each participant. The last participant last visit was on 11 January 2023. Participant disposition is shown in Fig. 1.

Patient disposition. EOS End of study. aPatients who initiated oral semaglutide treatment and attended the EOS visit. bPatients who were on oral semaglutide treatment and attended the EOS visit
The majority of participants were male (63.8%), and at baseline, participants had a mean (SD) age of 62 (10.4) years, duration of diabetes of 6.4 (5.3) years, HbA1c of 7.7 (1.5)%, body weight of 95.6 (17.9) kg and BMI of 33.2 (4.8) kg/m2 (Table 1). A total of 127 (68.6%) participants had a CV-related medical history (atrial fibrillation, chronic heart failure, coronary heart disease, hypertension, peripheral artery disease, revascularisation, stroke or transient ischaemic attack). Overall, 117 (63.2%) participants had a history of hypertension and 95 (51.4%) had a history of dyslipidaemia. Mean number of glucose-lowering medications at baseline was 0.8, with the most widely used being metformin (48.1% of participants), SGLT2is (13.0%) and sulphonylureas (7.0%). Overall, 56.2% of participants were receiving concomitant glucose-lowering medications at baseline.
Over half of all participants (54.1%) were treated by diabetes specialists in a secondary or tertiary care setting, with 45.9% treated by non-specialists in primary care. The most frequent reasons given for initiating oral semaglutide (multiple answers were possible) were to improve glycaemic control (87.0%), improve weight reduction (81.1%), address CV risk factors (35.7%) and simplify treatment regimen (8.6%). Almost all patients were initiated on the 3 mg dose (96.8%); however, five participants (2.7%) started on the 7 mg dose and one (0.5%) on the 14 mg dose.
Changes in HbA1c and Body Weight
There was a significant reduction in the primary endpoint of HbA1c from the observed baseline value of 7.8% to an estimated mean of 6.9% at EOS (in participants who had at least one post baseline HbA1c value and at EOS, n = 171), with an estimated change of − 0.91%-points (95% CI − 1.10, − 0.71; p < 0.0001), corresponding to − 9.89 mmol/mol (− 12.01, − 7.77; p < 0.0001) (Fig. 2a). Secondary and sensitivity analyses of change in HbA1c were consistent with the primary analysis (ESM Fig. S2).

Changes in a HbA1c (%, n = 171) and b body weight (kg, n = 174) from baseline to week 38. Data are from the in-study observation period. At week 0, observed mean at baseline for participants having at least one post-baseline assessment is plotted. Estimated change is analysed using an adjusted model, with baseline value, age, baseline BMI, time and time-squared as covariates, and sex, glucose-lowering agents at baseline, diabetes duration and site as fixed factors with random intercept and time (slope). The outer lines of the band represent the 95% CI. BL Baseline, BMI body mass index, EOS end of study, CI confidence interval, HbA1c glycated haemoglobin
Body weight also significantly decreased from baseline with an estimated absolute change of − 4.72 kg (95% CI − 5.60, − 3.84; p < 0.0001) and an estimated relative change of − 4.85% (95% CI − 5.70, − 4.00; p < 0.0001) at EOS (Fig. 2b). A post hoc analysis showed that change in body weight was independent from the type of clinic (primary vs. specialist) the participants were enrolled from (data not shown).
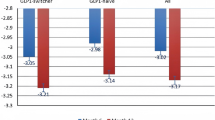
At EOS, 64.2% of participants had a HbA1c level < 7%, while 37.8% and 28.3% of participants achieved the composite endpoints of HbA1c reduction ≥ 1%-point plus body weight reduction ≥ 3% or ≥ 5% from baseline, respectively (Fig. 3). Post hoc analyses stratifying participants into subgroups based on baseline HbA1c (≤ 7%, > 7 to ≤ 8%, > 8 to ≤ 9% and > 9%) showed that while all subgroups had significant and clinically relevant reductions in HbA1c, even in those participants with the lowest baseline HbA1c (≤ 7%), participants in the higher HbA1c categories at baseline had numerically larger estimated reductions in HbA1c from baseline to end of study (ESM Tables S2, S3).

Secondary endpoints: proportion of participants achieving HbA1c targets and composite endpoints of HbA1c and body weight reduction at EOS. EOS End of study, HbA1c glycated haemoglobin. aNumber of participants with HbA1c < 7.0% at EOS was 137. bNumber of participants with HbA1c reduction ≥ 1 point and ≥ 3% or ≥ 5% body weight reduction at EOS was 127
Other Exploratory Endpoints
Mean waist circumference significantly decreased from baseline to EOS, with a change of − 4.54 cm (95% CI − 5.87, − 3.21; p < 0.0001). At EOS, 143 (77.3%) participants were being treated with oral semaglutide, among whom 94 (66.2%) participants were treated with the maximum 14 mg dose, 38 (26.8%) were on the 7 mg dose and 10 (7.0%) were receiving the 3 mg dose; one participant did not have their end dose recorded. More than a third of participants had changes to baseline concomitant glucose-lowering medication during the study period: 42 (22.7%) participants had an additional or increased dose of glucose-lowering medications, while 30 (16.2%) had ceased or received a decreased dose of glucose-lowering medication. At EOS, the mean number of glucose-lowering medications including oral semaglutide was 1.7, with metformin and SGLT2is the most frequent (62.2% and 16.2% of participants, respectively). A total of 57 (30.8%) participants were not receiving any concomitant glucose-lowering medications. Overall, treatment was considered a clinical success by treating physicians based on the individual targets set for 121 (73.8%) participants.
Safety
A total of 139 adverse events (AEs) were reported in 65 (35.1%) participants during the study. Most AEs were mild or moderate in severity (Table 2). The most frequent AEs were gastrointestinal disorders, which were reported by 50 (27.0%) participants. Safety concerns resulted in oral semaglutide being withdrawn in 22 (11.9%) participants; withdrawal in 20 patients (10.8%) was due to AEs, with gastrointestinal AEs (n = 19), primarily nausea (n = 12), accounting for all but one withdrawal. Oral semaglutide treatment was interrupted in 15 (8.1%) participants, and the dose reduced in 14 (7.6%) participants due to AEs. A total of three severe hypoglycaemic episodes were reported in one participant, who had taken only one medication, gliclazide, within 1 to 5 years before entering the study. This participant did not discontinue oral semaglutide, and completed treatment and the study. A total of six events in six (3.2%) participants were considered serious (COVID-19-related pneumonia, retinal detachment, radius fracture, urinary cystectomy, pyelonephritis and malignant melanoma progression), none of which were considered likely to be related to oral semaglutide. One participant died during the study due to malignant melanoma progression (also reported as a serious AE). This individual took oral semaglutide for 1 day; the death was reported 242 days after treatment with oral semaglutide and was considered unlikely to be related to treatment.
Discussion
The PIONEER REAL programme provides insights into how oral semaglutide is utilised in routine clinical practice in a diverse population of adult patients with T2D. These data indicate that people living with T2D treated with oral semaglutide in routine clinical practice in Switzerland achieve clinically significant improvements in glycaemic control and weight loss similar to those observed in clinical trials.
In this prospective, observational, real-world study conducted in primary and specialist care centres across Switzerland, participants living with T2D who had not previously been treated with injectable glucose-lowering drugs and who received oral semaglutide experienced clinically significant improvements in glycaemic control and body weight.
The decrease in HbA1c at EOS was estimated to be 0.91%-points in this study; of note, reductions of between 1.2- and 1.3%-points with oral semaglutide 14 mg after 52 weeks were reported in the PIONEER phase 3 programme (PIONEER 2, 3, 4, 7 and 8; based on the treatment policy estimand) [14, 15, 17,18,19]. The 0.91%-point reduction seen in the present analysis was similar to that observed in the real-world USA-based IGNITE study, in which HbA1c reduction was 0.9%-points after a mean treatment duration of 5.7 months (approximately 25 weeks) in people prescribed oral semaglutide [20]. The slightly lower HbA1c reduction observed in PIONEER REAL Switzerland compared with PIONEER trials might be attributable to the lower baseline HbA1c in participants in this study, which was 7.8% compared with 8.0–8.3% in the PIONEER trials [14, 15, 17, 18], although it should be noted that a post hoc analysis of PIONEER REAL Switzerland showed that participants with a higher HbA1c at baseline (32% of participants had HbA1c > 9% at baseline) achieved numerically larger reductions in HbA1c from baseline. Almost two-thirds of patients (n = 88 of 137, 64.2%) had HbA1c of < 7% at EOS, which is within the range seen at 52 weeks in PIONEER trials (63–72%) [14, 15, 17, 18]. The improvements in glycaemic control seen in this study are also in line with those reported with once-weekly subcutaneous semaglutide in the prospective real-world study, SURE Switzerland, in which participants had a HbA1c reduction of − 0.8%-points and 56% achieved HbA1c < 7.0% at EOS (approximately 30 weeks) [21].
Weight loss was 4.72 kg, corresponding to 4.85%, which was in line with the results reported after 52 weeks in PIONEER 2, 4, 7 and 8 (approximately 2.6–4.4 kg for oral semaglutide 14 mg, based on the treatment policy estimand) [14, 15, 17, 18] and similar to that reported with once-weekly subcutaneous semaglutide (5.0 kg corresponding to 5.0%) in the SURE Switzerland study [21].
Treatment was considered a clinical success by the physician in almost three-quarters of patients. However, it should be noted that clinical success is a subjective assessment and is dependent on individual targets set for each participant.
The PIONEER REAL studies provide insight on how oral semaglutide is prescribed in clinical practice. As would be expected based on the dosage information in the oral semaglutide label, almost all participants in PIONEER REAL Switzerland (96.8%) were initiated on the 3 mg dose. At EOS, 66.2% of participants were treated with the maximum currently approved dose of 14 mg and 26.8% were prescribed the 7.0 mg dose. A total of 10 participants (7.0%) were receiving the 3 mg dose, which is substantially lower than observed in IGNITE, in which 3 mg was received by 37% of participants [20]. However, the dosage results for oral semaglutide in PIONEER REAL Switzerland are consistent with those for once-weekly subcutaneous semaglutide in the SURE study and with those of oral semaglutide flexible dose in PIONEER 7 [17]. In the SURE Switzerland study, 8.6% of participants were on the lowest dose (0.25 mg) at EOS, 27.4% were receiving 0.5 mg and 61.7% were receiving the maximum dose (1.0 mg); in PIONEER 7, 9.0%, 30.2% and 59.4% of participants were receiving oral semaglutide 3, 7 and 14 mg [17, 21]. The reasons for the differences in the proportions of participants on each dose between the Swiss studies and IGNITE are unknown. One possible explanation for the use of a lower than maximum dose at EOS could be an increased occurrence, or perhaps expectation, of gastrointestinal AEs with higher doses. It is also possible that glycaemic targets were achieved at lower doses, discouraging the need for dose escalation. However, it is unknown whether participants who were on the 3 mg dose at study end had previously been on a higher dose or had stayed on 3 mg throughout the study, and due to the length of the study, participants may not have needed to escalate from the lowest dose of oral semaglutide.
The safety and tolerability of oral semaglutide were similar to that previously reported in phase 3 trials, with gastrointestinal effects, especially nausea and vomiting, being the most frequently reported AEs [13,14,15, 17, 18, 22, 23]. The discontinuation rate was similar to that seen in the PIONEER trials (7–13% with oral semaglutide 14 mg) [13,14,15, 17, 18, 22]. Reported discontinuation rates due to AEs were higher than observed in similar real-world studies with subcutaneous semaglutide, where rates of 4–5% were reported [21, 24]. However, in those studies, not all participants were treatment-naïve to GLP-1 RAs, and analyses were based on the on-treatment at EOS rather than the in-study population.
Over half of all participants in PIONEER REAL Switzerland were taking concomitant glucose-lowering medication at baseline, with metformin the most widely used. However, approximately 44% were not taking any concomitant glucose-lowering agents at baseline. The most recent recommendations of the Swiss Society for Endocrinology and Diabetes for the treatment of T2D advise first-line therapy with GLP-1 RA or SGLT2i in combination with metformin [5]. However, combination therapy involving a GLP-1 RA with a SGLT2i may not be reimbursed. Furthermore, in Switzerland, GLP-1 RA monotherapy is only reimbursed when metformin is not tolerated or contra-indicated, and the combination therapy of GLP-1 RA with metformin is only reimbursed when glycaemic control is insufficient with metformin alone. It should be noted that participants who were not taking other glucose-lowering agents at baseline were not necessarily treatment-naïve, as they may have previously been treated with glucose-lowering medications but had stopped before study enrolment, for example, due to side effects or perceived lack of effect.
The PIONEER REAL programme is the first prospective real-world evaluation of oral semaglutide. A key benefit of this study is that it involved the collection of data from a broad range of people living with T2D in Switzerland, with the cohort included in the study potentially more reflective of real-world practice and the results more generalisable than data from a more selected clinical trial population. The mean age of the cohort was 62 years with a high proportion of males, closely reflecting the population with treated T2D in Switzerland [25]. Limitations of this study include the observational design, the lack of a comparator arm and that data were collected as part of routine clinical practice. Consequently, the results are less robust than those based on randomised clinical trial data and potential unmeasured confounding factors cannot be excluded. In addition, unlike some other PIONEER REAL studies, the Swiss study did not include any assessment of patient satisfaction with treatment.
Compared with the SURE Switzerland study [21], the findings of PIONEER REAL Switzerland indicate that once-daily oral semaglutide offers similar glycaemic and weight loss benefits as the once-weekly subcutaneous formulation for people living with T2D that are treatment-naïve to injectable glucose lowering agents. As confirmed by a recent cross-sectional survey in nearly 4500 Italian patients [26], oral semaglutide represents an additional option that may increase acceptance and adherence compared with injectable formulations, reducing delays in treatment intensification (i.e. therapeutic inertia) and encouraging earlier use of GLP-1 RAs, including in primary care [27].
Conclusion
In conclusion, the PIONEER REAL Switzerland study demonstrates the use of once-daily oral semaglutide in a real-world setting to adults living with T2D in a Swiss population. The results achieved by the participants in this study presented here in both HbA1c measurements and body weight show that adults living with T2D can achieve significant improvements in clinical parameters related to T2D in a local clinical setting.
Data Availability
Data are available upon reasonable request. Data will be shared with bona fide researchers submitting a research proposal approved by the independent review board. Access request proposals can be found at novonordisk-trials.com. Data will be made available after research completion and approval of the product and product use in the European Union and the United States. Individual participant data will be shared in data sets in a de-identified/anonymised format.
References
Sun H, Saeedi P, Karuranga S, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119.
ElSayed NA, Aleppo G, Aroda VR, et al. 6. Glycemic targets: Standards of Care in Diabetes-2023. Diabetes Care. 2023;46:S97–110.
ElSayed NA, Aleppo G, Aroda VR, et al. 8. Obesity and weight management for the prevention and treatment of type 2 diabetes: Standards of Care in Diabetes-2023. Diabetes Care. 2023;46:S128–39.
Cosentino F, Grant PJ, Aboyans V, et al. 2019 ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J. 2020;41:255–323.
Gastaldi G, Lucchini B, Thalmann S, et al. Swiss recommendations of the Society for Endocrinology and Diabetes (SGED/SSED) for the treatment of type 2 diabetes mellitus (2023). Swiss Med Wkly. 2023;153:40060.
Ussher JR, Drucker DJ. Glucagon-like peptide 1 receptor agonists: cardiovascular benefits and mechanisms of action. Nat Rev Cardiol. 2023;20:463–74.
European Medicines Agency. Summary of product characteristics (Rybelsus® [semaglutide]). 2022. https://www.ema.europa.eu/en/medicines/human/EPAR/rybelsus. Accessed 14 Oct 2023.
US Food and Drug Administration. Highlights of prescribing information (Rybelsus® [semaglutide]). 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/213051s000lbl.pdf. Accessed 14 Oct 2023.
Swissmedic. Product Information (Rybelsus® [semaglutide]). 2023. https://www.swissmedicinfo.ch/ViewMonographie. Accessed 14 Oct 2023.
Swissmedic. Product information (Ozempic® [semaglutide]). 2023. https://www.swissmedicinfo.ch/ViewMonographie. Accessed 14 Oct 2023.
Swissmedic. Swiss Public Assessment Report (Ozempic® [semaglutide]). 2018. https://www.swissmedic.ch/swissmedic/en/home/humanarzneimittel/authorisations/new-medicines/semaglutidum.html. Accessed 14 Oct 2023.
Swissmedic. Swiss Public Assessment Report (Rybelsus® [semaglutide]). 2020. https://www.swissmedic.ch/swissmedic/en/home/about-us/publications/public-summary-swiss-par/public-summary-swiss-par-semaglutide.html. Accessed 14 Oct 2023.
Aroda VR, Rosenstock J, Terauchi Y, et al. PIONEER 1: randomized clinical trial of the efficacy and safety of oral semaglutide monotherapy in comparison with placebo in patients with type 2 diabetes. Diabetes Care. 2019;42:1724–32.
Rodbard HW, Rosenstock J, Canani LH, et al. Oral semaglutide versus empagliflozin in patients with type 2 diabetes uncontrolled on metformin: the PIONEER 2 trial. Diabetes Care. 2019;42:2272–81.
Pratley R, Amod A, Hoff ST, et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet. 2019;394:39–50.
Mosenzon O, Blicher TM, Rosenlund S, et al. Efficacy and safety of oral semaglutide in patients with type 2 diabetes and moderate renal impairment (PIONEER 5): a placebo-controlled, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7:515–27.
Pieber TR, Bode B, Mertens A, et al. Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): a multicentre, open-label, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7:528–39.
Zinman B, Aroda VR, Buse JB, et al. Efficacy, safety, and tolerability of oral semaglutide versus placebo added to insulin with or without metformin in patients with type 2 diabetes: the PIONEER 8 trial. Diabetes Care. 2019;42:2262–71.
Rosenstock J, Allison D, Birkenfeld AL, et al. Effect of additional oral semaglutide vs sitagliptin on glycated hemoglobin in adults with type 2 diabetes uncontrolled with metformin alone or with sulfonylurea: the pioneer 3 randomized clinical trial. JAMA. 2019;321:1466–80.
Aroda VR, Faurby M, Lophaven S, Noone J, Wolden ML, Lingvay I. Insights into the early use of oral semaglutide in routine clinical practice: the IGNITE study. Diabetes Obes Metab. 2021;23:2177–82.
Rudofsky G, Catarig AM, Favre L, et al. Real-world use of once-weekly semaglutide in patients with type 2 diabetes: results from the SURE Switzerland multicentre, prospective, observational study. Diabetes Res Clin Pract. 2021;178:108931.
Aroda VR, Aberle J, Bardtrum L, et al. Efficacy and safety of once-daily oral semaglutide 25 mg and 50 mg compared with 14 mg in adults with type 2 diabetes (PIONEER PLUS): a multicentre, randomised, phase 3b trial. Lancet. 2023;402:693–704.
Aroda VR, Erhan U, Jelnes P, et al. Safety and tolerability of semaglutide across the SUSTAIN and PIONEER phase IIIa clinical trial programmes. Diabetes Obes Metab. 2023;25:1385–97.
Yale JF, Catarig AM, Grau K, et al. Use of once-weekly semaglutide in patients with type 2 diabetes in routine clinical practice: results from the SURE Canada multicentre, prospective, observational study. Diabetes Obes Metab. 2021;23:2269–78.
Peytremann-Bridevaux I, Bordet J, Burnand B. Diabetes care in Switzerland: good, but perfectible: a population-based cross-sectional survey. BMC Health Serv Res. 2013;13:232.
Morieri ML, Candido R, Frontoni S, Disoteo O, Solini A, Fadini GP. Clinical features, cardiovascular risk profile, and therapeutic trajectories of patients with type 2 diabetes candidate for oral semaglutide therapy in the italian specialist care. Diabetes Ther. 2023;14:2159–72.
Evans M, Morgan AR, Bain SC, et al. Meeting the challenge of virtual diabetes care: a consensus viewpoint on the positioning and value of oral semaglutide in routine clinical practice. Diabetes Ther. 2022;13:225–40.
Medical Writing, Editorial, and Other Assistance.
The authors thank the study participants, investigators and study site staff who conducted the study. We also wish to thank Rahel Vogler, Medical Advisor, Novo Nordisk Pharma AG, for the set-up, initiation and conduct of the study. Medical writing support was provided by William Townley of Apollo, OPEN Health Communications, and Andy Bond, a contract writer working on behalf of Apollo, OPEN Health Communications, and funded by Novo Nordisk, in accordance with Good Publication Practice (GPP) guidelines (www.ismpp.org/gpp-2022).
Funding
This study was sponsored by Novo Nordisk A/S and is registered with ClinicalTrials.gov (NCT04537624). Novo Nordisk A/S also funded the rapid service fee of this manuscript.
Author information
Authors and Affiliations
Contributions
Hanan Amadid and Uffe Christian Braae contributed to study design and scientific operations. Anastas Kick, Flavio Acquistapace, Robert A. Ambühl, Bernd Schultes, Thomas Züger and Gottfried Rudofsky conducted the study and collected the data. Data were analysed by the sponsor. All authors participated in the interpretation of data, and revised the manuscript and approved the final version. All authors had full access to all the data in the study, actively contributed to all drafts of the manuscript and made the decision to submit the manuscript for publication.
Corresponding author
Ethics declarations
Conflict of Interest
Anastas Kick: no conflicts to disclose. Flavio Acquistapace: no conflicts to disclose. Robert A. Ambühl: no conflicts to disclose. Bernd Schultes: Bernd Schultes has received presentation fees from and compensation as a member of a scientific advisory board of Novo Nordisk and Eli Lilly, as well as for serving as a study investigator for Novo Nordisk. Thomas Züger: no conflicts to disclose. Gottfried Rudofsky: no conflicts to disclose. Khadija M'Rabet-Bensalah, Hanan Amadid, Flurin Item and Uffe Christian Braae are employees of and shareholders in Novo Nordisk.
Ethical Approval
The study protocol was approved by the appropriate health authorities according to local guidelines and by an Institutional Review Board/Independent Ethics Committee. The Ethics Committees included: Ethikkommission der Nordwest-und Zentralschweiz (EKNZ), Ethikkommission Zurich, Ethikkommission Ostschweiz (EKOS), Kantonale Ethikkommission Bern (KEK-Bern) and Commission cantonale (VD) d'éthique de la recherche sur l'être humain, Comitato etico cantonale Ticino. The study was conducted in accordance with the Declaration of Helsinki and International Council on Harmonisation Good Clinical Practice guidelines. Participants provided written informed consent prior to commencement of any study-related activity. The study is registered with ClinicalTrials.gov (NCT04537624).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kick, A., M’Rabet-Bensalah, K., Acquistapace, F. et al. Real-World Use of Oral Semaglutide in Adults with Type 2 Diabetes: The PIONEER REAL Switzerland Multicentre, Prospective, Observational Study. Diabetes Ther 15, 623–637 (2024). https://doi.org/10.1007/s13300-023-01525-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-023-01525-y