
ABSTRACT
Inter-individual heterogeneity in drug response is a serious problem that affects the patient’s wellbeing and poses enormous clinical and financial burdens on a societal level. Pharmacogenomics has been at the forefront of research into the impact of individual genetic background on drug response variability or drug toxicity, and recently the gut microbiome, which has also been called the second genome, has been recognized as an important player in this respect. Moreover, the microbiome is a very attractive target for improving drug efficacy and safety due to the opportunities to manipulate its composition. Pharmacomicrobiomics is an emerging field that investigates the interplay of microbiome variation and drugs response and disposition (absorption, distribution, metabolism and excretion). In this review, we provide a historical overview and examine current state-of-the-art knowledge on the complex interactions between gut microbiome, host and drugs. We argue that combining pharmacogenomics and pharmacomicrobiomics will provide an important foundation for making major advances in personalized medicine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Individual responses to a specific drug vary greatly in terms of both efficacy and toxicity. It has been reported that response rates to common drugs for the treatment of a wide variety of diseases fall typically in the range of 50%–75%, indicating that up to half of patients are seeing no benefit (Spear et al., 2001). Moreover, many individuals suffer from adverse drug reactions (ADRs). Approximately 3.5% of hospital admissions in Europe are related to ADRs, and about 10% of all patients in hospital experience an ADR during hospitalization (Bouvy et al., 2015). In the USA, serious drug toxicities cause over 100,000 deaths and cost 30–100 billion USD annually (Sultana et al., 2013). Inter-individual variability in drug response thus affects not only patient well-being, it also poses an enormous clinical and financial burdens. For the development of “personalized medicine”, it is therefore crucial to determine how we can assess a patient’s probable response to a drug, increase drug efficacy and reduce the risk of ADRs.
Over the past several decades, pharmacogenetics and pharmacogenomics have been at the forefront of research examining the impact of individual genetic make-up on drug response variability. It has now been estimated that genetic factors could explain 20%–95% of the variability in response to individual drugs (Kalow et al., 1998). Thus genetic factors alone are insufficient to explain the observed variability and other factors must be involved.
In recent years, the gut microbiota has emerged as an “organ” that plays an important role in health and disease. The human gut harbours thousands of different bacterial species and other microorganisms that form a complex ecosystem. The composition of the gut microbiome shows high inter-individual variation (Huttenhower and Human Microbiome Project Consortium, 2012) that is associated to a number of host and external factors (Falcony et al., 2016; Zhernakova et al., 2016). Various studies in mice and humans have shown the effect of drug intake on the gut microbiome (Forslund et al., 2015; Wu et al., 2017). In turn, the gut microbiome can also contribute to an individual’s response to a specific drug (Routy et al., 2018; Gopalakrishnan et al., 2017): the microbial community in the gut can modify the pharmacodynamics of a medication by directly transforming the drug or by altering the host’s metabolism or immune system.
Understanding the role of the gut microbiome in drug response may enable the development of microbiome-targeting approaches that enhance drug efficacy. The term pharmacomicrobiomics has been proposed to describe the influence of microbiome compositional and functional variations on drug action, fate and toxicity (Saad et al., 2012). Clearly, the gut microbiome is emerging as an essential component in the development of personalized medicine and modulating the gut microbiome has the potential to become a very attractive approach to managing drug efficiency and safety on the level of the individual.
THE COMPLEX SYSTEM OF DRUG METABOLISM
Orally ingested drugs may pass through the upper GI tract and small intestine into the large intestine, where they encounter the thousands of microbial species that reside in the human gut. Complex drug-microbial interactions occur mainly in the colon. Drugs may change intestinal microenvironment, alter microbial metabolism or affect bacterial growth, thereby altering microbial community composition and function. Conversely, the gut microbiome can also participate directly in chemical transformation of drugs (Fig. 1). In the host, drug metabolism occurs predominantly in the liver and can be divided into two phases of reactions: modification and conjugation. It has been noted, however, that the chemical modifications carried out by gut bacteria is very different from these hepatic processes. Gut microbes primarily conduct hydrolytic and reductive reactions to metabolize xenobiotics, while enzymes in the liver typically conduct oxidative and conjugative reactions (Koppel et al., 2017). Upon metabolism in the gut and/or the liver, drug metabolites are either transported to targeted tissues or excreted by the kidneys into the urine or by the liver via the biliary system back into the gut lumen. In the gut, drugs or their drug metabolites can be subjected again to bacterial metabolism (e.g., deconjugation) and (re)absorption (Stein et al., 2010). This complexity means that pharmacological studies require a systems biology approach that considers drug-related hepatic and bacterial metabolic processes, as well as complex host-microbe-drug interactions.

Sites and types of reactions for drug metabolism. Bacterial enzymes can participate in drug metabolism mainly through reductive and hydrolytic reactions. Drugs and their metabolites can be absorbed from the intestine and transported via the portal vein to the liver, where a fraction of them will be taken up and another fraction will spill over to the systemic circulation. Hepatic enzymes mainly conduct oxidative and conjugative reactions. Subsequently, drugs and/or their metabolites can be excreted back into the blood to be transported to targeted tissues, removed by the kidney via the urine, or directly excreted by the liver via the biliary system back into the gut lumen
MEDICATION PERTURBS THE GUT MICROBIOTA
Alteration in microbial composition and function by a drug can contribute to the overall effects of that drug on the host, which raises concerns in drug administration. Effects of antibiotics on the gut microbiome are the most studied, and antibiotic-induced dysbiosis in the gut microbiome can increase susceptibility to infections, compromise immune homeostasis, deregulate metabolism and obesity (Francino, 2016). Moreover, it is also a leading cause of Clostridium difficile infection, a severe intestinal inflammation caused by the overgrowing of this bacteria, which affects around 124,000 people per year and causes 3,700 deaths annually in Europe (European Centre for Disease Prevention and Control, 2015). Beyond antibiotics, a number of studies in humans and mice have now reported the impact of other commonly used drugs on the gut microbiome. This includes our metagenomics study in a Dutch population cohort of 1,135 samples, where we identified 19 drugs that affected gut microbiota composition (Zhernakova et al., 2016). A similar study in a Flemish cohort (FGFP cohort) reported that nearly 10% of inter-individual variation in the gut microbiome can be explained by medication use (Falcony et al., 2016). The medications identified in both studies were drugs prescribed for treatment of common diseases including gastro-oesophageal reflux, type II diabetes, depression, cardiovascular diseases and hyperlipidaemia.
While the majority of the current findings are association-based, the identification of a causal impact of proton pump inhibitors (PPIs), which are used to treat gastro-oesophageal reflux and heartburn, and the anti-diabetic drug metformin on gut microbiome composition provides firm evidence that alteration in gut microbiome should be considered when evaluating drug safety and that drug use can also confound microbiome analysis (Fig. 2A).

Drug-microbe effects. (A) Impact of drugs on the gut microbiome: drugs can perturb microbial composition and function. (B) Direct effect of gut microbiome on drug efficacy and toxicity: microbial transformation can activate or inactivate drugs, or induce drug toxicity to the host. (C) Indirect effect of gut microbiome on drug response: the gut microbiome can influence drug bioavailability and drug response via its interaction with host immune and metabolic systems. Specific examples illustrate each case
Proton pump inhibitors
PPIs are commonly used to treat acid-related diseases like gastro-oesophageal reflux disease. Acting through pH-dependent or pH-independent mechanisms, PPIs have the potential to alter the microbiota throughout different parts of the human gastrointestinal lumen (Freedberg et al., 2014). The impact of PPIs on the microbiome is widely reported (Imhann et al., 2016; Jackson et al., 2016). As PPIs reduce acidity in the stomach, there have been reports of overrepresentation of oral microbes in the gut (Imhann et al., 2016), likely due to a reduced stomach barrier function. This reduction in barrier function means pathogenic bacteria may also colonize the gut, and PPI users have a higher risk of enteric infections caused by Clostridium difficile (Dial et al., 2004). Interestingly, taxa alterations similar to those associated with C. difficile infection have also been seen in PPI users, including increased Streptococcus, Enterococcus and decreased Clostridiales (Freedberg et al., 2015). Another study showed that PPIs can accelerate endothelial senescence (Yepuri et al., 2016), although the role of gut microbiome in this adverse event remains unclear. Identification of the strong and unfavourable effect of PPIs on microbiome composition has led to discussions about banning their over-the-counter availability.
Metformin
Metformin is commonly used in the treatment of type II diabetes, and a beneficial impact of metformin in regulating the structure and function of the microbiota is emerging. Forslund et al. were the first to report that metformin could increase the abundance of bacteria that produce short chain fatty acids (SCFA), and these could mediate the therapeutic effects of metformin (Forslund et al., 2015). This observation was also confirmed by the observation of increased faecal levels of SCFAs in metformin users (Zhernakova et al., 2016). Metformin treatment has also been observed to increase the abundances of butyrate-producing bacteria and the mucin-degrading bacteria Akkermansia muciniphila (Forslund et al., 2015; Wu et al., 2017; Shin et al., 2014). Transferring human faecal samples from metformin-treated donors to germ-free mice improved glucose tolerance in the mice that received metformin-altered microbiota (Wu et al., 2017).
DIRECT IMPACTS OF THE GUT MICROBIOTA ON DRUG EFFICACY AND TOXICITY
Direct microbial effects on drug response are the chemical transformations of drug compounds by gut microbiota that influence a drug’s bioavailability or bioactivity and its toxicity (Koppel et al., 2017; Spanogiannopoulos et al., 2016) (Fig. 2B). To date, more than 30 drugs have been identified as substrates for intestinal bacteria (Jourova et al., 2016).
Recent insights into biotransformation of drugs by the gut microbiome and the clinical consequences hereof have led to a paradigm shift in pharmacokinetic analyses in humans.
Microbiome effects on drug activity
The first report of a microbial impact on drug activity can be dated back to 1930s with the discovery of the liberation of sulphanilamide via microbial transformation of prontosil (Fuller, 1937). Prontosil is an anti-bacterial drug and was one of a series of azo dyes examined by Gerhard Domagk for possible effects on haemolytic streptococcal infection, work for which he subsequently received the 1939 Nobel Prize in Medicine (Raju, 1999). It was subsequently observed that prontosil had no antibacterial action in vitro, and this led to the follow-up discovery in 1937 that its activity is due to the cleavage of the azo bond by bacterial azoreductases and the liberation of sulphanilamide that exerts anti-bacterial activity (Fuller, 1937). Sequentially, several prodrugs were developed with azo bonds that require bioactivation by gut microbes, including sulfasalazine, a drug in the treatment of ulcerative colitis. Bacterial cleavage of azo bonds in sulfasalazine in the intestine can favourably achieve site-specific release of the anti-inflammatory sulfapyridine and 5-aminosalicyclic acid (Peppercorn and Goldman, 1972).
Biotransformation by the gut microbiome can also inactivate drugs, as is seen with the drug digoxin. Digoxin is a commonly used cardiovascular drug, however, in around 10% of patients, the drug is converted to digoxin reduction products that are cardio-inactive. The inactivation of digoxin by gut microbiota was first reported in the 1980s, and antibiotic treatment resulted in a marked increase in serum concentrations of digoxin (Lindenbaum et al., 1981). The underlying mechanism remained unclear, however, until the discovery of specific Eggerthella lenta strains in 2013 (Haiser et al., 2013). By combining transcriptional profiling, comparative genomics and culture-based arrays, these E. lenta strains were identified as carrying a two-gene cardiac glycoside reductase (cgr) operon that is transcriptionally activated by digoxin. Arginine is proposed to serve as the main source of nitrogen and carbon for the growth of E. lenta (Sperry and Wilkins, 1976), while arginine could also inhibit digoxin inactivation (Saha et al., 1983). In line with this, the transcriptional activation of the cgr operon has been found to be dependent on arginine concentration. This observation has led to patients being encouraged to eat a high-protein-diet (high-arginine) to block inactivation of digoxin. More recently, Kumar et al. found that the binding pocket of digoxin at the cgr operon primarily involves negatively charged polar amino acids and a few non-polar hydrophobic residues and fumarate, which can bind to the same binding sites but with a higher binding energy than digoxin. This knowledge may lead to development of drugs that block cgr binding sites (Kumar et al., 2017).
Microbiome effects on drug toxicity
Toxicity occurs when the bacterial transformation of a drug leads to the generation of metabolites that have harmful effects on the host. The role of the gut microbiome on chemotherapy efficacy and toxicity has been recently well discussed (Alexander et al., 2017). One of the best known examples involves the bacterial enzyme β-glucuronidases, which has been described to be involved in the toxicity of the common colon cancer chemotherapeutic CPT-11 (also known as irinotecan). Up to 80% patients using CPT-11 can present with severe diarrhoea. CPT-11 is primarily metabolized in the liver, where human carboxylesterases first activate CPT-11 to its cytotoxic metabolite SN-38, which then inhibits the nuclear topoisomerase 1 enzyme critical for DNA replication. In drug elimination, SN-38 is glucuronidated to its inactive form SN-38G by the liver UDP-glucuronosyltransferase (UGT). SN-38G is excreted via the biliary track into the gut, where bacterial β-glucuronidases can re-activate the drug by converting SN-38G back to SN-38 (Stein et al., 2010), which exhibits toxicity toward intestinal epithelial cells and causes diarrhoea. Via the same mechanism, bacterial β-glucuronidases can also induce toxicity of non-steroidal anti-inflammatory drugs (NSAIDs), which can cause gastroduodenal mucosal lesions in up to 50% of users (Higuchi et al., 2009). When glucuronidated NSAIDs secreted via the hepatobiliary pathway reach the distal small intestinal lumen, bacterial β-glucuronidases produce aglycones that can be taken up by enterocytes. Intestinal cytochrome P450s further metabolize aglycones to potentially reactive intermediates that induce severe endoplasmic reticulum stress or mitochondrial stress leading to cell death (Boelsterli et al., 2013). This mechanism explains the toxic effect of NSAIDs on the intestinal wall.
Because β-glucuronidases are present in a wide range of dominant gut bacteria (Dabek et al., 2008), it is a challenge to design bacterium-specific targets to reduce drug toxicity. Yet modulating activity of bacterial enzymes has become an attractive approach to alleviate drug toxicity. Wallace et al. have identified several β-glucuronidase inhibitors that can efficiently inhibit enzyme activities in living aerobic and anaerobic bacteria while not affecting bacterial growth or harming host epithelial cells (Wallace et al., 2010). Mouse experiments have now shown that oral administration of inhibitors efficiently alleviates drug toxicity of CPT-11, pointing to a potential drug to reduce β-glucuronidase-driven drug toxicity (Wallace et al., 2010).
INDIRECT IMPACT OF THE GUT MICROBIOTA ON DRUG RESPONSE
Indirect microbial effects are microbial influences on drug bioavailability and response via an impact of their metabolic or peptide products on the host immune system or host metabolism (Wu et al., 2017; Gopalakrishnan et al., 2017; Routy et al., 2018) (Fig. 2C). One representative example of microbial impact on drug bioavailability is seen for simvastatin, a drug commonly prescribed in the treatment of hyperlipidaemia. Plasma concentrations of simvastatin are positively associated with microbially synthesized secondary bile acids (Kaddurah-Daouk et al., 2011). Since bile acids are important agents for intestinal nutrient absorption, this also may determine the absorption of simvastatin into the host and influence the drug’s bioavailability.
A very intriguing example of microbial impact on drug response can be seen in recent advances in immunotherapy efficacy (Gopalakrishnan et al., 2017; Routy et al., 2018). In oncology, one of the most promising anti-cancer therapies is immunotherapy aimed at alleviation of the blockade of immune checkpoints, using treatments including PD-1/PD-L1 blockers or anti-CTLA4 therapy (Fig. 3A). However, response to these therapies is often heterogeneous. The influence of the gut microbiome on immunotherapy response was first reported in mice by Sivan et al. (2015), who found that commensal Bifidobacterium showed a positive association with antitumor T cell response and Bifidobacterium-treated mice showed a significant improvement in tumour control. This mouse study led to a follow-up study in human cancer patients (Gopalakrishnan et al., 2017) comparing gut microbial composition in 112 melanoma patients undergoing anti-PD-1 therapy. It showed that patients responding to immunotherapy had higher gut microbial diversity and a higher abundance of Clostridiales, Ruminococcaceae and Faecalibacterium. This microbiome structure may enhance systemic and anti-tumour immune response via increased antigen presentation and improved effector T cell function. In contrast, the non-responders had lower microbial diversity and a higher abundance of Bacteroidales. Another independent study in patients with epithelial tumours also showed that individual response to PD-l/PD-L1 blockers is determined by gut microbiome composition (Routy et al., 2018). In drug responders, over-representation was observed for Akkermansia, Ruminococcus spp., Alistipes spp. and Eubacterium spp., while under-representation was found for Bifidobacterium adolescentis, B. longum and Parabacteroids distasonis. This study further explored potential microbiome-modulating therapeutic approaches to enhance drug response and found that simply avoiding antibiotics while taking PD-1 blockers could boost the patient’s positive response from the current 25% up to 40%. Probiotics, like orally administrated A. muciniphila, can also enhance the response to PD-1 blockers in humans and mice. While further study is still needed to elucidate the underlying mechanisms, it is plausible that this beneficial effect is exerted via the anabolic functions of the gut microbiome, which may promote host immunity. For instance, SCFAs, a major type of bacterial metabolites from dietary fibres, could influence differentiation of T-helper 17 (Th17) and T-regulatory (Treg) cells (Omenetti and Pizarro, 2015). SCFAs have also been implicated in anti-inflammatory properties of Clostridia strains (Atarashi et al., 2013; Smith et al., 2013), which would have a beneficial effect on immune function and epithelial permeability (Stefka et al., 2014). Moreover, the gut microbiome has been found to determine an individual’s inflammatory cytokine production in response to different pathogens (Schirmer et al., 2016).

Gut microbiome associated with response of PD-1/PD-L1 based immunotherapy. (A) Checkpoints of immunotherapy. Programmed cell death protein 1 (PD-1) is a cell surface receptor that serves as an immune checkpoint. This receptor plays an important role in suppressing T cell inflammatory activity and down-regulating the immune system. Tumour cells can express PD-1 ligands (PD-L1) that are able to bind to PD-l protein and thus inactivate T cells. Accordingly, several PD-1/PD-L1 blockers have been designed to block the interaction between PD-1 and PD-L1 to enable anti-tumour immunity. (B) Gut microbes associated with individual response of immunotherapy and the proportion of their variation explained by host genetics and environmental factors. Five bacterial taxa are associated to higher response, while bacteroidales is linked to low response. Inter-individual variation is scaled as 1 and the proportion of explained variation by genetic factors and environmental factors are shaded blue and green, respectively. Estimation of explained variation derived from the TwinsUK study: Goodrich et al. (2014) Cell, 159:789–799
COMPLEX GENETICS-DIET INTERACTION IN PHARMACOMICROBIOMICS
Pharmacomicrobiomic studies that aim to investigate the bidirectional effects between the gut microbiome and drugs need to consider that the gut microbiome is itself a complex trait. It can be affected by host genetics, exogenous factors and by their interactions. Genome-wide association studies (GWAS) have shown that the microbial composition in an individual’s gut can be affected by genetic variants involved in innate immunity, metabolism and food processing. Exogenous factors like diet also have marked effects on the gut ecosystem: what we eat also feeds our gut microbes. A western diet and lifestyle (i.e., a high calorie, high fat diet and a sedentary lifestyle) is widely reported to be associated with a less diverse microbial ecology than, for instance, a high fibre diet. In addition, we have reported associations of 68 dietary factors to the gut microbiome (Zhernakova et al., 2016). However, our diet also contains bioactive compounds that can interact with drugs. Therefore, there is a tripartite interaction between genetics, the gut microbiome and exogenous factors (including diet) in drug metabolism (Fig. 4).

Host-microbe-diet interactions in drug metabolism. Complex drug-microbe interactions can result in alterations in microbial composition and function and change the chemical structure of compounds that could directly or indirectly affect drug metabolism in the liver. Moreover, genetics and exogenous factors, including diet, can affect both gut microbiome and drug metabolism in the host
Impact of host genetics on gut microbiome and drug metabolism
Once a drug is administered, it interacts with targets (such as transporters, receptors and enzymes), may undergo metabolism, and is then removed from the system. Each of these processes could potentially involve clinically significant genetic variants (Touw, 1997). Over the past decades, pharmacogenetic studies have identified numerous genetic variants via GWAS (Spear et al., 2001). For instance, the toxicity of CPT-11 discussed above is not only linked to the gut microbiome (Stein et al., 2010) but also to genetic variants in the UGT1A1 gene. Around ~10% of the western population carries a genetic variant in UGT1A1 that leads to poor drug metabolism and results in a higher risk for severe toxicity (Innocenti et al., 2004). Clinical decision-making has been increasingly incorporating information of human genetic variation. It has also been shown that individual tailored drug administration based on genotype of CYP2C9 and VKORC1 genes can reduce risk of hospitalization caused by the commonly used anticoagulant warfarin by as much as 30% (Madian et al., 2012).
The impact of host genetics on the gut microbiota has also been emerging. Benson et al. conducted the first QTL-based association study in mouse intercross lines and provided clear evidence for the impact of genetic variation on the gut microbiome (Benson et al., 2010). The heritability of individual bacteria in humans was first estimated in 416 pairs of twins from the TwinsUK cohort (Goodrich et al., 2014). The host genetic influence on the gut ecosystem might also have an impact on microbes involved in drug toxicity and efficacy. If we focus, for instance, on taxa associated with the modulation of PD-1/PD-L1 blocker response in immunotherapy, we see that a large proportion are heritable, for example, Bifidobacterium (h2 = 0.32), Ruminococcaceae (h2 = 0.20), Faecalibacterium (h2 = 0.18), A. muciniphila (h2 = 0.12) (Fig. 3B).
Several GWAS have been conducted in humans to identify individual genetic variants associated to gut microbiome in humans (Goodrich et al., 2014; Bonder et al., 2016; Goodrich et al., 2016; Wang et al., 2016). Despite limited direct overlap of associated loci across different studies, a difference potentially due to different analysis methods and low statistical power, the associated loci from all studies generally converge into several physiological processes involved in innate immunity, metabolism and food processing. In particular, C-type lectin molecules and functional variants in the lactase gene (LCT) have been consistently associated to gut microbiome composition and pathways in several studies (Kurilshikov et al., 2017). This not only highlights the complex host-microbe immune and metabolic interactions, it also provides an opportunity to study the causal role of the gut microbiome in health and disease by using genetic variants as instrumental variables in causal inference analysis (a Mendelian randomization approach) (Sheehan et al., 2008).
Impact of diet on gut microbiome and drug metabolism
Dietary components are extrinsic factors that can affect various physiological processes in humans and gut microbial composition. Many microbial enzymes involved in drug metabolism can also metabolize dietary components. Drug-diet interaction occurs when the consumption of a particular food affects the absorption of a drug or modulates the activity of drug-metabolizing enzymes, resulting in altered pharmacokinetics of the drug. The impact of dietary protein and fat on drug metabolism was first noted in the 1970s (Campbell and Hayes, 1976). With the progress of pharmacogenomics, more insights have been obtained at the molecular level. For instance, some drug-metabolizing enzymes have been found to be very sensitive to dietary effects, including several members of Cytochrome P450 family (CYP3A4, CYP1A2 and CYP2E1) and P-glycoprotein (P-gp) transporters localized at the gut epithelium (Harris et al., 2003; Markowitz et al., 2003; Peters et al., 2016). It has been suggested that dietary factors can alter expression levels of these enzymes/transporters in the intestine, as well as their substrate-specificity, thus affecting drug metabolism by these enzymes.
Diet is one of the most important exogenous factors shaping the gut microbiome. Long-term and short-term effects of dietary factors on the gut microbiome are well-documented (Falcony et al., 2016; Zhernakova et al., 2016; Rothschild et al., 2017; David et al., 2013). Understanding the impact of diet on microbiome-mediated drug metabolism can identify dietary covariates to be corrected for in microbiome analysis and indicate the potential of tailored dietary advice during drug treatment to enhance drug efficacy. One example already in practice is the high-protein diet suggested during digoxin treatment to block inactivation of digoxin by E. lenta (Haiser et al., 2013). A. muciniphila not only enhances the response rate of PD-1/PD-L1 blockers but also exerts beneficial effect on metabolic health. High abundance of A. muciniphila is associated to low BMI, low risk of type II diabetes and a healthy lipid profile (Dao et al., 2016; Plovier et al., 2017; Everard et al., 2013). A. muciniphila has been identified as a mucin-degrading bacterium that resides in the mucus layer and that abundantly colonizes in nutrient-rich environments (Derrien et al., 2004). Inter-individual variations in A. muciniphila are mostly linked to environmental factors (Fig. 3B) (Goodrich et al., 2014). It has been reported that dietary polyphenols can promote the growth of A. muciniphila in mice (Roopchand et al., 2015) and that administration of oligofructose to genetically obese mice increased the abundance of A. muciniphila by ~100-fold (Everard et al., 2011). Moreover, metformin is found to increase SCFA-producing bacteria in the human gut, which can contribute to the therapeutic effects of metformin (Forslund et al., 2015; Wu et al., 2017). However, production of SCFAs also requires dietary fibres. Indeed, a recent dietary intervention study has revealed that weight loss in metformin users is positively associated with higher dietary fibre intake but not with total carbohydrate intake (Sylvetsky et al., 2017). Thus, certain dietary components or prebiotics can induce shifts in the gut microbiome and thereby modulate drug responses. However, we also should bear in mind that such effects can be bi-directional as drugs can also perturb the gut microbiome. In addition, the gut microbiome may also determine an individual’s response to dietary interventions (Zeevi et al., 2015).
SYSTEMS PROSPECTIVE IN PHARMACOGENOMICS AND PHARMACOMICROBIOMICS
With the complex diet-drug-host-microbe interaction in mind, a great challenge lies ahead of us in predicting an individual’s response to a specific drug. This is central for successful implementation and clinical application of personalized (or precision) medicine. Two successful applications thus far are the Israeli personalized nutrition study, which predicted individual postprandial glycaemic response using a machine-learning algorithm to integrate blood parameters, dietary habits, anthropometrics, physical activity and gut microbiome (Zeevi et al., 2015), and the drug prediction algorithm, vedoNet, which incorporates microbiome and clinical data to predict the individual response to IBD treatment (Ananthakrishnan et al., 2017). To move further toward clinical applications, it is important to understand the underlying causality and mechanisms, an aim which require a systems biology approach coupling pharmacogenetics, pharmacogenomics and pharmacomicrobiomics to improve our understanding of factors that control drug pharmacokinetics at the individual level.
Well-characterized human cohorts
An “ideal” systems biology study in humans should facilitate the generation of data from the same individuals over time on multiple dimensional levels. It should include information on diet and lifestyle, living environment, presence of disease and use of drugs, and incorporate multiple omics layers via information on genetics, transcriptome, proteome, metabolome and gut microbiome. A number of large biobanks that capture gut microbiome data have now been established, including the LifeLines-DEEP cohort (Zhernakova et al., 2016; Tigchelaar et al., 2015), the UK biobank and TwinsUK cohort (Goodrich et al., 2014; Sudlow et al., 2015), the Flemish cohort (Falcony et al., 2016) and the Israeli personalized nutrition cohort (Zeevi et al., 2015). These cohorts have greatly advanced our understanding of host-microbe interactions in health and disease and of their interplay with exogenous factors. Preferably, the cohorts should have a longitudinal design in order to tackle perturbations, perform interventions and predict disease outcomes based on factors such as genetic risk, gut microbiome, molecular biomarkers, physiological traits and environmental factors. The longitudinal, prospective LifeLines cohort, for example, has been following 167,000 individuals for 10 years and will continue to do so for another 20 years. Within LifeLines, questionnaires on lifestyle, disease, drug use, quality of life, and other factors are collected regularly, and all participants undergo physical measurements every 5 years, with fasting biological samples (blood, urine, etc.) being collected at the same time. Over 2,000 phenotypic factors are recorded for each individual (Scholtens et al., 2015). In addition to this large collection of physiological and lifestyle factors, several initiatives have been set up to generate deep molecular data to enable systems biology studies (Fig. 5). For example, the LifeLines-DEEP cohort, a subset of 1,500 individuals from the LIfeLines cohort, is deeply profiled for various “omics” data on the genome, epigenome, transcriptome, proteome, metabolome and gut microbiome (Tigchelaar et al., 2015). LifeLines-DEEP serves as the foundation for systems biology and systems genetics analyses and for understanding inter-individual variation in the gut microbiome and host-microbe interaction in health and disease (Zhernakova et al., 2016; Imhann et al., 2016; Bonder et al., 2016; Fu et al., 2015; Tigchelaar et al., 2016). In recent years, two additional initiatives have been launched. One is the LifeLines DAG3 study, which is collecting oral, airway and gut microbiome data from 10,000 individuals across a wide age range (8–91 years). Metagenomic sequencing of the LifeLines DAG3 samples is underway to assess taxonomy, strain diversity and functionality. Uniquely, not only are all individuals fully genotyped but glycerol aliquots of their microbiota are also stored to enable bacterial culture for further functional studies. LifeLines DAG3 will have the power to study host-microbe interactions and will also allow studies to move from association to causality. The second initiative is the LifeLines NEXT cohort, which includes 1,500 pregnant women and their newborns and performs detailed phenotyping across the first year of life to study the development and maturation of the gut microbiome and virome and the impact of genetics and environmental factors on the developing microbial ecosystem. In all three initiatives, genetics, gut microbiome, medication use, diseases, dietary and environmental factors are available for each individual. This offers a great opportunity to systematically investigate individual variability in drug metabolism and underlying genetic, microbial, dietary factors and their interactions.

Overview of LifeLines-DEEP cohort. LifeLines-DEEP is a subset of 1,500 individuals from the large, prospective, population-based LifeLines cohort (n = 167,000 individuals). In addition to information about >2,000 exogenous factors (morphological, physiological, clinical), LifeLines-DEEP participants have been deeply profiled for multiple “omics” data layers
Moving from association to causality. Population-based studies with deep omics data provide powerful means for identifying risk factors in humans. Several bioinformatics-based causal inference methods, including Mendelian randomization approaches and structural equation modelling, can be used. However, this inferred causality requires further experimental validation. Transplanting the whole microbiome, specific species or a mixture into model organisms has proven to be a powerful method to illustrate causality. For instance, transferring human microbiota to germ-free mice has validated the causal role of the microbiome in mediating therapeutic effects of metformin and autoimmune therapy (Forslund et al., 2015; Wu et al., 2017; Routy et al., 2018). However, it is increasingly clear that animal models fall short in predicting pharmacokinetics in humans. It is well established that gut microbiome, metabolism and drug responses are very different between mouse and human. The knowledge generated in mouse models, even in humanized mouse models, is not directly applicable to humans. Thus several studies have conducted clinical interventions in humans to prove causality of interactions in humans, as shown by the effect of metformin (Wu et al., 2017). Yet, due to the obvious practical and ethical issues associated with human clinical studies, very little causality has actually been validated in humans. New approaches that allow for individualized drug testing in vitro may be set to change this landscape.
CULTUROMICS AND ORGAN-ON-CHIP: NEW OPPORTUNITIES FOR DEVELOPING PERSONALIZED TREATMENTS
Individualized drug testing in vitro is urgently needed as a tool to aid precision medicine. Such a model should also be able to trace chemical transformation of drugs and mimic mechanical, structural, absorptive, transport and pharmaceutical properties of drugs within the gut-liver system. Given the interplay between the gut microbiome and host genome in drug metabolism, there is increasing awareness that we should take both personal microbial composition and personal genome into account when considering personalized medicine.
Microfluidic organs-on-chips are an emerging technology that mimics human organs or tissues. Differentiating human induced pluripotent stem cells (iPSCs) can give rise to different cell types that carry the genetic makeup of the iPSC-donors. An individual’s iPSCs can be programmed to become gut epithelial cells or liver hepatocytes using FGF/BMP-induced differentiation. This innovative and non-invasive technology enables functional study in the milieu of an individual’s genetic background, which holds great promise in disease-modelling and drug-testing applications (Bhatia and Ingber, 2014; Huh et al., 2011). The potential of this approach has been shown in various applications using organoid models and organ-on-chips (Trietsch et al., 2017; Kim et al., 2012; Takayama et al., 2012; Takebe et al., 2014). For instance, a liver-on-a-chip has been engineered that mimics heterotypic interaction by separating iPSC-derived hepatocytes from the active flow microchannels designed to resemble the natural endothelial barrier of the liver sinusoid (Huh et al., 2011). The device has now been proven to maintain metabolic activity of the hepatocytes for over 7 days and to permit metabolic analysis.
Most gut microbes are strictly anaerobic and, for much of the last century, fewer than 30% of them could be cultured in the laboratory, which made functional studies impossible. With advances in culture-independent next-generation sequencing technologies, we have started to gain more insights into the composition and function of gut microbes based on their DNA sequencing. These bioinformatics-based approaches have yielded many insights into the underlying mechanisms. However, to further validate these mechanisms requires functional studies using a culture-based approach. In recent years bacterial culture technologies have been developed that now allow around 80% of gut microbes to be cultured (Lagier et al., 2016), making functional validation of gut microbe processes finally possible. For instance, bacterial metabolism of corticosteroids and ranitidine has been determined using in vitro cultures (Yadav et al., 2013; Basit and Lacey, 2001). These cultures attempt to closely mimic the colonic environment for specific strains of bacteria and host-microbe interactions. Drugs are then added to assess their impact on bacterial growth and metabolism and, vice versa, how bacteria chemically transform these drugs.
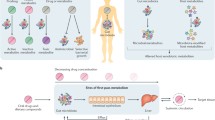
With the advance of organs-on-chips and the bacterial culturomics, we anticipate that in vitro models in the next-phase of personalized medicine will also be able to couple personalized genome and metagenome and to conduct individual-based drug testing on cultured bacteria, gut epithelial cells and hepatocytes simultaneously (Fig. 6). We will thus be able to apply drug breakdown products of bacterial enzymes or other bacterial metabolites to the gut-on-a-chip and liver-on-a-chip to further investigate their effect on the host cells. Moreover, drug metabolites produced by hepatic enzymes can also be applied to the gut microbiome and gut epithelial cells to evaluate whether their enzyme can re-activate drugs and cause adverse events.

Individual-based drug testing. Advance of bacterial “culturomics”, development of organs-on-chip and high-throughput metabolism and pharmacokinetic analyses, will enable individual-based in vitro drug testing in the near future. For this purpose, liver gut microbiome can be collected for culturing. This can be done either on whole community level or on individual species or strain level (blue arrows). Currently, organs-on-chips are emerging as a next-generation drug-testing model system. Non-invasive collection of urine leads to human induced pluripotent stem cells from which we can generate different types of tissue cells (e.g., gut epithelial cells or hepatocytes) (green arrows). These cells will have exactly the same genetic background. Coupling cultured bacteria and organs-on-chip offers a high potential to conduct individual-based drug testing, by taking into consideration both an individual’s own genome and his/her metagenome (red arrows)
CONCLUSIONS
The microbes residing in the human gut encode a broad diversity of enzymes, greatly expanding the repertoire and capacity of metabolic reactions in the human body that can be involved in xenobiotic metabolism, including that of dietary components and drugs. The gut microbiome is thus emerging as an important player in personalized medicine. Several papers have discussed the role of the gut microbiome on drug efficacy and toxicity (Alexander et al., 2017). Here, we are considering the host-microbe-drug interactions and the impact of dietary factors. We further propose a comprehensive analysis framework that combines well-characterized human cohorts and innovative in vitro model systems to study this complex interaction. Notably, in contrast to human genetic make-up, gut microbiota can be modulated. Pharmacomicrobiomics thus may have at least two global clinical applications: 1) to combine personal microbiome and genetic profiles to better predict an individual’s medication response; and 2) to modulate the gut microbiome to improve drug efficacy on the individual level. However, given the great diversity of microbial composition, its broad function in the host and the complex drug-diet-microbe-host interactions, a systems-based approach and individualized drug testing systems are needed to further understand the underlying causalities and mechanisms. The recent advance in cutting-edge, state-of-art technologies in bacterial culturomics and individualized organs-on-chips, together with exponential growth of databanks and biobanks holding vast amounts of information about the same individual, will enable the development of the next phase in personalized medicine.
References
Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM (2017) Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol 14:356–365
Ananthakrishnan AN, Luo C, Yajnik V, Khalili H, Garber JJ, Stevens BW, Cleland T, Xavier RJ (2017) Gut microbiome function predicts response to anti-integrin biologic therapy in inflammatory bowel diseases. Cell Host Microbe 21:603.e3–610.e3
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K et al (2013) Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500:232–236
Basit AW, Lacey LF (2001) Colonic metabolism of ranitidine: implications for its delivery and absorption. Int J Pharm 227:157–165
Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K et al (2010) Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA 107:18933–18938
Bhatia SN, Ingber DE (2014) Microfluidic organs-on-chips. Nat Biotechnol 32:760–772
Boelsterli UA, Redinbo MR, Saitta KS (2013) Multiple NSAID-induced hits injure the small intestine: underlying mechanisms and novel strategies. Toxicol Sci 131:654–667
Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP et al (2016) The effect of host genetics on the gut microbiome. Nat Genet 48:1407–1412
Bouvy JC, De Bruin ML, Koopmanschap MA (2015) Epidemiology of adverse drug reactions in Europe: a review of recent observational studies. Drug Saf 38:437–453
Campbell TC, Hayes JR (1976) The effect of quantity and quality of dietary protein on drug metabolism. Fed Proc 35:2470–2474
Dabek M, McCrae SI, Stevens VJ, Duncan SH, Louis P (2008) Distribution of beta-glucosidase and beta-glucuronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol Ecol 66:487–495
Dao MC, Everard A, Aron-Wisnewsky J, Sokolovska N, Prifti E, Verger EO, Kayser BD, Levenez F, Chilloux J, Hoyles L et al (2016) Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 65:426–436
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA et al (2013) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563
Derrien M, Vaughan EE, Plugge CM, de Vos WM (2004) Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol 54:1469–1476
Dial S, Alrasadi K, Manoukian C, Huang A, Menzies D (2004) Risk of Clostridium difficile diarrhea among hospital inpatients prescribed proton pump inhibitors: cohort and case–control studies. CMAJ 171:33–38
European surveillance of Clostridium difficile infections. Surveillance protocol version 2.2 (2015) European Centre for Disease Prevention and Control. https://doi.org/10.2900/44795
Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Muccioli GM, Neyrinck AM, Possemiers S, Van Holle A, François P et al (2011) Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60:2775–2786
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM et al (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110:9066–9071
Falcony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles-Colomer M, Vandeputte D et al (2016) Population-level analysis of gut microbiome variation. Science 352:560–564
Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Krogh Pedersen H et al (2015) Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528:262–266
Francino MP (2016) Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front Microbiol. https://doi.org/10.3389/fmicb.2015.01543
Freedberg DE, Lebwohl B, Abrams JA (2014) The impact of proton pump inhibitors on the human gastrointestinal microbiome—clinical key. Clin Lab Med 34:771–785
Freedberg DE, Toussaint NC, Chen SP, Ratner AJ, Whittier S, Wang TC, Wang HH, Abrams JA (2015) Proton pump inhibitors alter specific taxa in the human gastrointestinal microbiome: a crossover trial. Gastroenterology 149:883–885
Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JAM, Brandsma E, Marczynska J, Imhann F, Weersma RK et al (2015) The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ Res 117:817–824
Fuller AT (1937) Is p-aminobenzenesulphonamide the active agent in protonsil therapy? Lancet (Lond) 229:194–198
Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT et al (2014) Human genetics shape the gut microbiome. Cell 159:789–799
Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, Spector TD, Bell JT, Clark AG, Ley RE (2016) Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19:731–743
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC et al (2017) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 4236:1–15
Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ (2013) Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 341:295–298
Harris RZ, Jang GR, Tsunoda S (2003) Dietary effects on drug metabolism and transport. Clin Pharmacokinet 42:1071–1088
Higuchi K, Umegaki E, Watanabe T, Yoda Y, Morita E, Murano M, Tokioka S, Arakawa T (2009) Present status and strategy of NSAIDs-induced small bowel injury. J Gastroenterol 44:879–888
Huh D, Hamilton GA, Ingber DE (2011) From 3D cell culture to organs-on-chips. Trends Cell Biol 21:745–754
Huttenhower C, Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214
Imhann F, Bonder MJ, Vila AV, Fu J, Mujagic Z, Vork L, Tigchelaar EF, Jankipersadsing SA, Cenit MC, Harmsen HJM et al (2016) Proton pump inhibitors affect the gut microbiome. Gut 65:740–748
Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, Karrison T, Janisch L, Ramírez J, Rudin CM et al (2004) Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol 22:1382–1388
Jackson MA, Goodrich JK, Maxan M-E, Freedberg DE, Abrams JA, Poole AC, Sutter JL, Welter D, Ley RE, Bell JT et al (2016) Proton pump inhibitors alter the composition of the gut microbiota. Gut 65:749–756
Jourova L, Anzenbacher P, Anzenbacherova E (2016) Human gut microbiota plays a role in the metabolism of drugs. Biomed Pap 160:317–326
Kaddurah-Daouk R, Baillie RA, Zhu H, Zeng ZB, Wiest MM, Nguyen UT, Wojnoonski K, Watkins SM, Trupp M, Krauss RM (2011) Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS ONE. https://doi.org/10.1371/journal.pone.0025482
Kalow W, Tang B-K, Endrenyi L (1998) Hypothesis: comparisons of inter- and intra-individual variations can substitute for twin studies in drug research. Pharmacogenetics 8:283–289
Kim HJ, Huh D, Hamilton G, Ingber DE (2012) Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 12:2165
Koppel N, Rekdal VM, Balskus EP (2017) Chemical transformation of xenobiotics by the human gut microbiota. Science 356:1246–1257
Kumar K, Jaiswal SK, Dhoke GV, Srivastava GN, Sharma AK, Sharma VK (2017) Mechanistic and structural insight into promiscuity based metabolism of cardiac drug digoxin by gut microbial enzyme. J Cell Biochem. https://doi.org/10.1002/jcb.26638
Kurilshikov A, Wijmenga C, Fu J, Zhernakova A (2017) Host genetics and gut microbiome: challenges and perspectives. Trends Immunol 38:633–647
Lagier J-C, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P, Caputo A, Cadoret F, Traore SI, Seck EH et al (2016) Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol 1:16203
Lindenbaum J, Rund DG, Butler VPJ, Tse-Eng D, Saha JR (1981) Inactivation of digoxin by the gut flora: reversal by antibiotic therapy. N Engl J Med 305:789–794
Madian AG, Wheeler HE, Jones RB, Dolan ME (2012) Relating human genetic variation to variation in drug responses. Trends Genet 28:487–495
Markowitz JS, Donovan JL, DeVane CL, Taylor RM, Wort TJOHNS, Per HY, Ruan Y, Wang J-S, Chavin KD (2003) Effect of St John’s wort on drug metabolism by induction of cytochrome P450 3A4 enzyme. JAMA 290:1500–1504
Omenetti S, Pizarro TT (2015) The Treg/Th17 axis: a dynamic balance regulated by the gut microbiome. Front Immunol. https://doi.org/10.3389/fimmu.2015.00639
Peppercorn MA, Goldman P (1972) The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther 181:555–562
Peters SA, Jones CR, Ungell AL, Hatley OJD (2016) Predicting drug extraction in the human gut wall: assessing contributions from drug metabolizing enzymes and transporter proteins using preclinical models. Clin Pharmacokinet 55:673–696
Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, Chilloux J, Ottman N, Duparc T, Lichtenstein L et al (2017) A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med 23:107–113
Raju TN (1999) The nobel chronicles. Lancet 353:681
Roopchand DE, Carmody RN, Kuhn P, Moskal K, Rojas-Silva P, Turnbaugh PJ, Raskin I (2015) Dietary polyphenols promote growth of the gut bacterium Akkermansia muciniphila and attenuate high-fat diet-induced metabolic syndrome. Diabetes 64:2847–2858
Rothschild D, Weissbrod O, Barkan E, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, Zmora N et al (2017) Environmental factors dominate over host genetics in shaping human gut microbiota composition. bioRxiv. https://doi.org/10.1101/150540
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP et al (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359:91–97
Saad R, Rizkallah MR, Aziz RK (2012) Gut pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog 4:16
Saha JR, Butler VP, Neu HC, Lindenbaum J (1983) Digoxin-inactivating bacteria: identification in human gut flora. Science 220:325–327
Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Jansen T, Jacobs L, Bonder MJ, Kurilshikov A et al (2016) Linking the human gut microbiome to inflammatory cytokine production capacity. Cell 167:1125.e8–1136.e8
Scholtens S, Smidt N, Swertz MA, Bakker SJL, Dotinga A, Vonk JM, Van Dijk F, Van Zon SKR, Wijmenga C, Wolffenbuttel BHR et al (2015) Cohort profile: LifeLines, a three-generation cohort study and biobank. Int J Epidemiol 44:1172–1180
Sheehan NA, Didelez V, Burton PR, Tobin MD (2008) Mendelian randomisation and causal inference in observational epidemiology. PLoS Med 5:e177
Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, Lee MS, Bae JW (2014) An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63:727–735
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Man Lei Y, Jabri B, Alegre M-L et al (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350:1084–1089
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569–573
Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ (2016) The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol 14:273–287
Spear BB, Heath-Chiozzi M, Huff J (2001) Clinical application of pharmacogenetics. Trends Mol Med 7:201–204
Sperry J, Wilkins T (1976) Arginine, a growth-limiting factor for Eubacterium lentum. J Bacteriol 127:780–784
Stefka AT, Feehley T, Tripathi P, Qiu J, McCoy K, Mazmanian SK, Tjota MY, Seo G-Y, Cao S, Theriault BR et al (2014) Commensal bacteria protect against food allergen sensitization. Proc Natl Acad Sci USA 111:13145–13150
Stein A, Voigt W, Jordan K (2010) Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management. Ther Adv Med Oncol 2:51–63
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M et al (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. https://doi.org/10.1371/journal.pmed.1001779
Sultana J, Cutroneo P, Trifirò G (2013) Clinical and economic burden of adverse drug reactions. J Pharmacol Pharmacother 4:S73–S77
Sylvetsky AC, Edelstein SL, Walford G, Boyko EJ, Horton ES, Ibebuogu UN, Knowler WC, Montez MG, Temprosa M, Hoskin M et al (2017) A high-carbohydrate, high-fiber, low-fat diet results in weight loss among adults at high risk of type 2 diabetes. J Nutr. https://doi.org/10.3945/jn.117.252395
Takayama K, Inamura M, Kawabata K, Katayama K, Higuchi M, Tashiro K, Nonaka A, Sakurai F, Hayakawa T, Furue MK et al (2012) Efficient generation of functional hepatocytes from human embryonic stem cells and induced pluripotent stem cells by HNF4α transduction. Mol Ther 20:127–137
Takebe T, Zhang RR, Koike H, Kimura M, Yoshizawa E, Enomura M, Koike N, Sekine K, Taniguchi H (2014) Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat Protoc 9:396–409
Tigchelaar EF, Zhernakova A, Dekens JAM, Hermes G, Baranska A, Mujagic Z, Swertz MA, Muñoz AM, Deelen P, Cénit MC et al (2015) Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open 5:e006772
Tigchelaar EF, Bonder MJ, Jankipersadsing SA, Fu J, Wijmenga C, Zhernakova A (2016) Gut microbiota composition associated with stool consistency. Gut 65:540–542
Touw DJ (1997) Clinical implications of genetic polymorphisms and drug interactions mediated by cytochrome P-450 enzymes. Drug Metab Drug Interact 14:55–82
Trietsch SJ, Naumovska E, Kurek D, Setyawati MC, Vormann MK, Wilschut KJ, Lanz HL, Nicolas A, Ng CP, Joore J et al (2017) Membrane-free culture and real-time barrier integrity assessment of perfused intestinal epithelium tubes. Nat Commun. https://doi.org/10.1038/s41467-017-00259-3
Wallace BD, Hongwei W, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh L-A, Mani S et al (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330:831–835
Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, Hov JR, Degenhardt F, Heinsen F-A, Rühlemann MC, Szymczak S et al (2016) Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet 48:1396–1406
Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, Ståhlman M, Olsson LM, Serino M, Planas-Fèlix M et al (2017) Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 23:850–858
Yadav V, Gaisford S, Merchant HA, Basit AW (2013) Colonic bacterial metabolism of corticosteroids. Int J Pharm 457:268–274
Yepuri G, Sukhovershin R, Nazari-Shafti TZ, Petrascheck M, Ghebre YT, Cooke JP (2016) Proton pump inhibitors accelerate endothelial senescence. Circ Res 118:e36–e42
Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben-Yacov O, Lador D, Avnit-Sagi T, Lotan-Pompan M et al (2015) Personalized nutrition by prediction of glycemic responses. Cell 163:1079–1095
Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G, Vieira-Silva S et al (2016) Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352:565–569
ACKNOWLEDGEMENTS
M. Doestzada holds a MD–PhD Fellowship from University Medical Center Groningen. A. Zhernakova is funded by a Netherlands Organization for Scientific Research (NWO) VIDI Grant (016.178.056) and a European Research Council (ERC) Starting Grant (715772). A. Zhernakova also holds a Rosalind Franklin Fellowship (University of Groningen). R. K. Weersma is funded by an NWO VIDI (016.136.308). D. J. Touw is funded by ZonMw (Grant No. 80-83600-98-42014), Astellas, Chiesi and the Tekke Huizinga Fund. C.W. is funded by an ERC Advanced Grant (FP/2007-2013/ERC Grant 2012-322698), NWO Spinoza Prize (92-266), Stiftelsen Kristian Gerhard Jebsen Foundation (Norway), NWO Gravitation Grant Netherlands for the Organ-on-Chip Initiative (024.003.001) and University of Groningen Investment Agenda Grant for Personalized Health. J. Fu is funded by an NWO-VIDI (864.13.013). A. Zhernakova, D. P. Y. Koonen, F. Kuipers and J. Fu are also supported by CardioVasculair Onderzoek Nederland (CVON 2012-03). We thank Kate Mc Intyre for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Doestzada, M., Vila, A.V., Zhernakova, A. et al. Pharmacomicrobiomics: a novel route towards personalized medicine?. Protein Cell 9, 432–445 (2018). https://doi.org/10.1007/s13238-018-0547-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-018-0547-2