
ABSTRACT
The loss of or decreased functional pancreatic β-cell is a major cause of type 1 and type 2 diabetes. Previous studies have shown that adult β-cells can maintain their ability for a low level of turnover through replication and neogenesis. Thus, a strategy to prevent and treat diabetes would be to enhance the ability of β-cells to increase the mass of functional β-cells. Consequently, much effort has been devoted to identify factors that can effectively induce β-cell expansion. This review focuses on recent reports on small molecules and protein factors that have been shown to promote β-cell expansion.
Similar content being viewed by others

INTRODUCTION
The prevalence of the diabetic population in the United States is 29.1 million or 9.3% of the total population as shown in a recent National Diabetes Statistics Report (CDC, 2014). In China, the overall prevalence of diabetes is 11.6% in the adult population (Xu et al., 2013). Clearly diabetes is becoming a serious worldwide health problem as currently 350 million individuals in the world suffer from diabetes (Vetere et al., 2014). The lack of functional pancreatic β-cells leads to diabetes. Therefore, a better understanding of how an appropriate number of functional β-cells is generated and maintained shall help develop strategies for diabetic treatment. Three major approaches for increasing β-cell mass involve induction of β-cell proliferation, enhancement of β-cell viability and β-cell reprogramming (Vetere et al., 2014). This review intends to focus on regulation of β-cell proliferation. While some studies indicate that adult β-cell replication or neogeneration was hard to detect (Gunasekaran et al., 2012; Guardado-Mendoza et al., 2012; Cavelti-Weder et al., 2013; Xiao et al., 2013a, b), an increase of β-cell mass has been reported under a non-diabetic obesity condition (Klöppel et al., 1985), and in other studies in animals (Hull et al., 2005; Bock et al., 2003) and in humans (Heit et al., 2006; Rahier et al., 2008; Hanley et al., 2010; Saisho et al., 2013). During pregnancy, an increase of β-cell mass was also observed, which produced more insulin to set off insulin resistance (Sorenson and Brelje, 1997; Toselli et al., 2014). Moreover, adult β-cell proliferation was found in pancreas impaired through pancreatectomy and partial duct ligation (Dor et al., 2004; Peshavaria et al., 2006; Nir et al., 2007; Xiao et al. 2013a, b). Therefore, adult β-cells appear to be still capable of proliferation. Finding ways to enhance such capacity to expand β-cell mass shall provide important strategies for diabetic treatment.
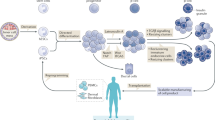
Previous studies have led to the identification of many chemical compounds and biological factors that can increase β-cell mass and some recent reviews have looked into β-cell proliferation control through intracellular signaling (e.g. Kulkarni et al., 2012; Bernal-Mizrachi et al., 2014). This review focuses on small molecules and protein factors that are known to have an effect on the induction of β-cell expansion but their mechanisms of action still require further investigations (Fig. 1).

Several factors summarized in this review act through intracellular pathways to elicit β-cell expansion by enhancing cell proliferation and/or reducing cell death. In the case of FTY720, its effect on β-cells appears to be mediated through activation of immune cells in the lymph nodes. For the most part, molecular mechanisms of these pathways remain to be fully elucidated. GABA: γ-aminobutyric acid; OC: osteocalcin; OEA: oleoylethanolamide; S1P: sphingosine-1-phosphate; TEF: trefoil factor; TH: thyroid hormone; GPCR: G protein-coupled receptor. Gprc6a: the G protein-coupled receptor family C group 6 protein; GK: glucokinase. GK activators are small chemical compounds
γ-AMINOBUTYRIC ACID
The γ-aminobutyric acid (GABA) is a product derived from the amino acid glutamate (Fenalti et al., 2007). Extracellular glutamate enters β-cells through the glutamate transporter-1 (GLT-1), and is then converted to GABA (Adeghate and Ponery, 2002). GABA can be secreted by β-cells and acts as an autocrine through a positive feedback loop in pancreatic β-cells (Braun et al., 2010). It is reported that inflammation can increase β-cell proliferation (Sherry et al., 2006), and GABA could inhibit inflammation which probably partially counteracted its ability to induce proliferation (Song and Park, 2014). However, Tian et al. (2013) observed that GABA increased β-cell mass through enhanced survival and proliferation of β-cells. In rodents, GABA increased β-cell proliferation and suppressed apoptosis through up-regulated BCL-XL and dampened Caspase-3 (Ligon et al., 2007). Excitingly, it has been reported that GABA restores β-cell mass and reverse diabetes in GABA-injected mice (Soltani et al., 2011).
THYROID HORMONE
The relationship between iodothyronine and diabetes has been studied since 1980’s. Decreased serum T3 was observed after insulin was withdrawn in juvenile type diabetic patients (Madsbad et al., 1981). It was reported that thyroid hormones reduce glucose tolerance both in animals and in humans (Lenzen and Bailey, 1984). To determine whether thyroid hormones play a role in β-cell proliferation, Ximenes et al. (2007) reported that high concentration of T3 (>250 µmol/L) attenuated β-cell proliferation, increased apoptosis and decreased the secretion of insulin, while physiological concentrations of T3 have no negative effects on the function and survival of β-cells. However, more studies have shown that thyroid hormone plays a positive role in regulating β-cells mass. T3 has been shown to reduce the risk of type 1 diabetes in autoimmune prone BB rats (Hartoft-Nielsen et al., 2009). Furthermore, T3 treatment increased β-cell mass in Wistar rats. T3 has also been shown to induce the proliferation of β-cell through the MAPK pathway and enhance the secretion of insulin (Kim et al., 2014). Aguayo-Mazzucato et al. (2013) reported that thyroid hormone promotes β-cell development through Mafa, which is a key transcription factor of β-cell differentiation (Kaneto et al., 2008; 2009). Rat pancreatic β-cell lines (RIN5F) treated with T3 induced cell proliferation, and further research shows the Cyclin D1/CDK/Rb/E2F pathway was involved in the process of β-cell replication. Furthermore, intra pancreatic injection of TRα led to an expansion of the β-cell mass in diabetic mice (Furuya et al., 2013). Moreover, ligand-bound thyroid hormone receptor TRα is involved in the reprogramming of pancreatic acinar cells into insulin-producing cells via activation of PI3K signaling (Furuya et al., 2013).
TREFOIL FACTORS
Trefoil factors 2/3 (TFF2 and TFF3) are members of the trefoil family which are expressed in gastrointestinal mucosa. TFF3 is expressed in fetal human and newborn rat pancreas. Strong expression of TFF3 was found in islets and some pancreatic duct cells. There was significant expression of TFF3 mRNA in human islet samples examined. Exogenous TFF3 promoted islet cell attachment and migration, but had no effect on proliferation (Jackerott et al., 2006). However, over-expression of TFF3 in a RIN cell line and rat islets using recombinant adenovirus lead to an increased proliferation which can be arrested by knockdown of TFF3 using siRNA. Mediated by SDF-1α/CXCR4 signaling, another family member TFF2 promotes cell proliferation through increasing ERK1/2 phosphorylation in rat INS-1 cells, mouse MIN6 cells, and mouse islets (Orime et al., 2013). As TFF2 is not expressed in pancreatic islets (Jackerott et al., 2006), circulating TFF2 may enhance pancreatic β-cell expansion by interacting with its receptor. The serum TFF2 levels are reportedly increased during pregnancy (Samson et al., 2008; 2011). Thus, TFF2 might contribute to pancreatic β-cell expansion during pregnancy.
OSTEOCALCIN
Osteocalcin (OC) is a Vitamin K-dependent protein secreted in the late differentiation stage of osteoblasts. It has been considered as an endocrine hormone because of its ability to reduce insulin sensitivity, decrease fat mass, and induce release of glucagon-like peptide-1 and thereby stimulate insulin secretion (Lee et al., 2007; Ferron et al., 2008; Mizokami et al., 2013). Through a molecular genetic approach, Wei et al (2014) have recently found that osteocalcin stimulates β-cell proliferation in the pancreas via a Cyclin D1-dependent mechanism utilizing the G protein-coupled receptor family C group 6 protein (Gprc6a). This stimulation occurs during the peak of β-cell proliferation, which occurs in the perinatal period, and in adult mice. Moreover, they described the effects of daily osteocalcin injections in obese type 2 diabetic mice, reporting an increase in the number of mitochondria in skeletal muscles and an increase in energy expenditure. It indicates that osteocalcin can increase muscle work by increasing insulin sensitivity. Elucidation of how OC/Gprc6a signaling promotes β-cell proliferation may provide a novel approach for diabetes treatment (Wei et al., 2014).
GPR119 AND S1P RECEPTOR
G protein-coupled receptor 119 (GPR119) is expressed in the pancreas in rodents and humans. A selective small molecular GPR-119 agonist, PSN632408, can reduce food intake and body weight gain in rodents through increasing intracellular cAMP levels (Overton et al., 2006). PSN632408 and an endogenous ligand of GPR119, oleoylethanolamide (OEA), can stimulate β-cell replication in mouse islets in vitro. OEA and PSN632408 improved mouse islet graft function in diabetic mouse insulin-positive/BrdU-positive β-cells. OEA and PSN632408 treatment increased active GLP-1 levels in mice plasma (Gao et al., 2011). The ability of PSN632408 to stimulate β-cell replication in cultured mouse islets and in vivo has been recently demonstrated (Ansarullah et al. 2013).
Many of the immune suppressive drugs are toxic to β-cells. Because of this, their clinical administration after islet transplantation for type 1 diabetes was limited. However, Truong and collegues found that FTY720, an immune suppressor that modulates sphingosine-1-phosphate receptor (S1PR, a G protein-coupled receptor) activity, did not impair human islet function in vitro or in vivo (Truong et al., 2007). Interestingly, treatment with FTY720 can prevent the onset of diabetes in an animal model of human type 1 diabetes by activating immune cells in the lymph nodes (Jörns et al., 2010). Moreover, oral administration of FTY720 to obese mice can increase β-cell mass and blood insulin levels. This function is mediated by decreasing the cyclin-dependent kinase inhibitor p57 (KIP2) level, and at the same time, increasing the cyclin D3 level (Zhao et al., 2012). By inhibiting β-cell apoptosis, FTY720 can retain β-cell mass and prevent damage of pancreatic islet (Moon et al., 2013). Through finding ultra-structural changes in pancreatic β-cells after treatment with anti-TCR and FTY720 in type 1 diabetic rats, a similar improvement of β-cell viability has been observed (Jörns et al., 2014).
GLUCOKINASE ACTIVATORS
Glucokinase (GK) activator is effective in lowering blood glucose concentration not only by the enhancement of glucose uptake in the liver but also by the secretion of insulin from pancreatic β-cell (Park, 2012). Activation of glucokinase by small chemical compound promotes pancreatic β-cell proliferation. When treated with GKA50, a GK agonist, INS-1 β-cell proliferation increased at basal levels of glucose. This effect is mediated by the IRS-2/PI3K/PKB pathway. Moreover, GKA50 was found to prevent INS-1 cell apoptosis under the impairment of chronic high glucose conditions (Wei et al., 2009). YH-GKA, another GK activator, also increased the INS-1 β-cell number by up-regulating IRS-2 and subsequently activating AKT/PKB. IRS-2 down-regulation can decrease the proliferation effect of YH-GKA. YH-GKA induces ATP content and citrate synthase activity which blocks β-cell apoptosis (Oh et al., 2014). Importantly, GKA was shown to be sufficient and effective in promoting β-cell proliferation in mice (Salpeter et al., 2010). Positive impact of GK agonists on promoting β-cell proliferation and preserving β-cell mass has been shown in aging mice and diabetic rat models (Stolovich-Rain et al., 2012; Futamura et al., 2012).
OTHER FACTORS
Betatrophin has been shown to induce β-cell proliferation in a mouse model of insulin resistance on the basis of gain-of-function evidence derived from over-expression of betatrophin in the mouse liver (Yi et al., 2013). Betatrophin is mainly expressed in the liver and fat and its plasma level is associated with β-cell proliferation in insulin resistance mice and the mouse model during gestation. Treatment with an insulin receptor antagonist S961 elicited insulin resistance and led to an incremental quantity of betatrophin. Blocking the insulin receptor with a high dose of S961 led to the mice glucose intolerant and an increase in β-cell replication. However, a recent study using both betatrophin knockout and over-expression approaches indicate that betatrophin does not control β-cell expansion (Gusarova et al., 2014). β-Cells from individual mice appear to have a broad range of responses to betatrophin and redundancy may exist that could compensate for the loss of betatrophin function (Yi et al., 2014). In any event, further investigation is needed to clarify whether betatrophin can indeed play a role in regulating β-cell expansion together with other factors.
Some other factors have been reported to be able to induce β-cell proliferation. Early studies revealed that a lectin from Agaricus bisporus (mushroom) (ABL) causes a dose-dependent inhibition of tumor cell proliferation (Yu et al., 1993; 1999). Surprisingly, a recent research found that ABL administration promoted β-cell proliferation (Wang et al., 2012). It is unclear how ABL has opposite effects on tumor cell and β-cell proliferation.
The IGF-1 receptor (IGF1R) has become a therapeutic target for cancer treatment. The efficacy of OSI-906, a dual inhibitor of IGF1R and insulin receptor, was found to elicit β-cell proliferation to increase β-cell mass in male mice (Shirakawa et al., 2014). While insulin signaling in β-cells was not affected by OSI-906, how OSI-906 treatment leads to β-cell expansion needs to be further investigated.
Using a high throughput primary β-cell replication assay, two adenosine kinase (ADK) inhibitors, 5-Iodotubercidin and ABT-702, have been identified and shown to increase β-cell mass (Annes et al., 2012).
Finally, a unique compound epoxypukalide was reported to induce a 2.5-fold increase in β-cell proliferation, through activation of the ERK1/2 signaling pathway and up-regulation of Cyclin D2/Cyclin E. Epoxypukalide did not attenuate glucose-stimulated insulin secretion in rat islets (López-Acosta et al., 2013). The mechanism of epoxypukalide action remains to be elucidated.
CONCLUDING REMARKS
Until now, there is no promising medication for expanding β-cell mass for diabetic treatment. However, strong evidence has been accumulated to support that β-cell proliferation could be enhanced by small chemical compounds or extracellular factors in animal models (Fig. 1). As β-cell proliferation was detected in a surgically resected pancreas from an 89-year-old with recent-onset diabetes (Meier et al., 2006), this observation encourages further exploration of strategies to promote adult β-cell expansion as a therapeutic approach for treatment of diabetes.
Clearly much more efforts are needed to identify small molecules and protein factors that can explicitly elicit human pancreatic β-cell regeneration. Along this line, it is important to keep in mind that significant differences exist between human and other animals such as mice and rat. Therefore, tests must be further conducted with β-cells in cultured human pancreatic cell lines or islet tissues if a factor is initially discovered and studied in animals. Moreover, any potential therapeutic factors should be able to effectively target β-cells in vivo to minimize possible side effects.
Because cell proliferation is typically regulated through intercellular signaling, many growth factors and hormones have been tested for their ability to influence β-cell proliferation. For example, osteocalcin produced by osteoblasts in the bone acts as a hormone to stimulate β-cell proliferation in the pancreas (Wei et al., 2014). Similarly, there are other hormones such as prolactin and thyroid hormone increased in gestation, and leptin increased in adiposity to elicit β-cell proliferation.
While it is challenging to identify novel substances that increase β-cell mass, efforts have been made to test some existing medications such as antioxidants, immune-suppressants, and even anti-cancer drugs, for their effect on β-cell expansion. Interestingly, an inhibitor for both IGF receptor and insulin receptor can effectively expand β-cell mass (Shirakawa et al., 2014). It remains to be clarified as to how this inhibitor of IGF/insulin signaling, a potent growth-promoting pathway, acts to enhance β-cell proliferation and survival. Not surprisingly, most of these drugs exhibited shortcomings with regards to their specificity and efficiency.
Currently, most of the β-cell research focuses on individual factors for their effect on β-cell expansion. However, β-cell expansion occurs in a complex patho-physiological background and therefore, multiple factors should be considered to simultaneously interfere with several steps or pathways to enhance β-cell proliferation. For example, a recently discovered novel hormone, irisin, was deemed to cross-talk with betatrophin in the process of β-cell regeneration and dedifferentiation (Boström et al., 2012; Sanchis-Gomar and Perez-Quilis, 2014; Zhang et al., 2014). It would be interesting to learn how such inter-molecular and inter-pathway interactions affect β-cell expansion.
Notably, β-cells can proliferate while retaining their differentiated phenotypes in vivo. This fact provides another facet to reflect the complexity of β-cell proliferation in vivo. Given all that, in the future we might need a “cocktail” program to realize the efficiency and safety of β-cell regeneration for dealing with diabetes. Although we are facing a hard situation, obviously, we are on the right path.
Abbreviations
- ADK:
-
adenosine kinase
- GABA:
-
γ-aminobutyric acid
- GK:
-
Glucokinase
- GLT-1:
-
glutamate transporter-1
- OC:
-
osteocalcin
- OEA:
-
oleoylethanolamide
- TFF2/3:
-
trefoil factors 2/3
REFERENCES
Adeghate E, Ponery AS (2002) GABA in the endocrine pancreas: Cellular localization and function in normal and diabetic rats. Tissue Cell 34:1–6
Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, Hollister-Lock J, El Khattabi I, Marsili A, Weir GC, Sharma A, Larsen PR, Bonner-Weir S (2013) Thyroid hormone promotes postnatal rat pancreatic β-cell development and glucose-responsive insulin secretion through MAFA. Diabetes 62:1569–1580
Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA (2012) Adenosine kinase inhibition selectively promotes rodent and porcine islet β-cell replication. Proc Natl Acad Sci USA 109:3915–3920
Ansarullah, Lu Y, Holstein M, DeRuyter B, Rabinovitch A, Guo Z (2013) Stimulating β-cell regeneration by combining a GPR119 agonist with a DPP-IV inhibitor. PLoS One 8:e53345
Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocaña A (2014) Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes 63:819–831
Bock T, Pakkenberg B, Buschard K (2003) Increased islet volume but unchanged islet number in ob/ob mice. Diabetes 52:1716–1722
Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Boström EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Højlund K, Gygi SP, Spiegelman BM (2012) A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481:463–468
Braun M, Ramracheya R, Bengtsson M, Clark A, Walker JN, Johnson PR, Rorsman P (2010) GABA is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 59:1694–1701
Cavelti-Weder C, Shtessel M, Reuss JE, Jermendy A, Yamada T, Caballero F, Bonner-Weir S, Weir GC (2013) Pancreatic duct ligation after almost complete β-cell loss: exocrine regeneration but no evidence of β-cell regeneration. Endocrinology 154:4493–4502
Centers for Disease Control and Prevention (2014) National diabetes statistics report: estimates of diabetes and its burden in the United States, 2014. U.S. Department of Health and Human Services, Atlanta
Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429:41–46
Fenalti G, Law RH, Buckle AM, Langendorf C, Tuck K, Rosado CJ, Faux NG, Mahmood K, Hampe CS, Banga JP, Wilce M, Schmidberger J, Rossjohn J, El-Kabbani O, Pike RN, Smith AI, Mackay IR, Rowley MJ, Whisstock JC (2007) GABA production by glutamic aciddecarboxylase is regulated by a dynamic catalytic loop. Nat Struct Mol Biol 14:280–286
Ferron M, Hinoi E, Karsenty G, Ducy P (2008) Osteocalcin differentially regulates β cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci USA 105:5266–5270
Furuya F, Shimura H, Asami K, Ichijo S, Takahashi K, Kaneshige M, Oikawa Y, Aida K, Endo T, Kobayashi T (2013) Ligand-bound thyroid hormone receptor contributes to reprogramming of pancreatic acinar cells into insulin-producing cells. J Biol Chem 288:16155–16166
Futamura M, Yao J, Li X, Bergeron R, Tran JL, Zycband E, Woods J, Zhu Y, Shao Q, Maruki-Uchida H, Goto-Shimazaki H, Langdon RB, Erion MD, Eiki J, Zhou YP (2012) Chronic treatment with a glucokinase activator delays the onset of hyperglycaemia and preserves beta cell mass in the Zucker diabetic fatty rat. Diabetologia 55:1071–1080
Gao J, Tian L, Weng G, Bhagroo NV, Sorenson RL, O’Brien TD, Luo J, Guo Z (2011) Stimulating beta cell replication and improving islet graft function by GPR119 agonists. Transpl Int 24:1124–1134
Guardado-Mendoza R, Jimenez-Ceja L, Majluf-Cruz A, Kamath S, Fiorentino TV, Casiraghi F, Velazquez AO, DeFronzo RA, Dick E, Davalli A, Folli F (2012) Impact of obesity severity and duration on pancreatic β-cell and α-cell dynamics in normoglycemic non-human primates. Int J Obes 37:1071–1078
Gunasekaran U, Hudgens CW, Wright BT, Maulis MF, Gannon M (2012) Differential regulation of embryonic and adult β cell replication. Cell Cycle 11:2431–2442
Gusarova V, Alexa CA, Na E, Stevis PE, Xin Y, Bonner-Weir S, Cohen JC, Hobbs HH, Murphy AJ, Yancopoulos GD, Gromada J (2014) ANGPTL8/Betatrophin does not control pancreatic beta cell expansion. Cell 159:691–696
Hanley SC, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, Petropavlovskaia M, Rosenberg L (2010) Beta-cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology 151:1462–1472
Hartoft-Nielsen ML, Rasmussen AK, Bock T, Feldt-Rasmussen U, Kaas A, Buschard K (2009) Iodine and tri-iodo-thyronine reduce the incidence of type 1 diabetes mellitus in the autoimmune prone BB rats. Autoimmunity 42:131–138
Heit JJ, Karnik SK, Kim SK (2006) Intrinsic regulators of pancreatic beta-cell proliferation. Annu Rev Cell Dev Biol 22:311–338
Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE (2005) Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 48:1350–1358
Jackerott M, Lee YC, Møllgård K, Kofod H, Jensen J, Rohleder S, Neubauer N, Gaarn LW, Lykke J, Dodge R, Dalgaard LT, Søstrup B, Jensen DB, Thim L, Nexø E, Thams P, Bisgaard HC, Nielsen JH (2006) Trefoil factors are expressed in human and rat endocrine pancreas: differential regulation by growth hormone. Endocrinology 147:5752–5759
Jörns A, Rath KJ, Terbish T, Arndt T, Meyer Zu, Vilsendorf A, Wedekind D, Hedrich HJ, Lenzen S (2010) Diabetes prevention by immunomodulatory FTY720 treatment in the LEW.1AR1-iddm rat despite immune cell activation. Endocrinology 151:3555–3565
Jörns A, Akin M, Arndt T, Terbish T, Zu Vilsendorf AM, Wedekind D, Hedrich HJ, Lenzen S (2014) Anti-TCR therapy combined with fingolimod for reversal of diabetic hyperglycemia by β cell regeneration in the LEW.1AR1-iddm rat model of type 1 diabetes. J Mol Med 92:743–755
Kaneto H, Miyatsuka T, Kawamori D, Yamamoto K, Kato K, Shiraiwa T, Katakami N, Yamasaki Y, Matsuhisa M, Matsuoka TA (2008) PDX-1 and MafA play a crucial role in pancreatic beta-cell differentiation and maintenance of mature beta-cell function. Endocr J 55:235–252
Kaneto H, Matsuoka TA, Katakami N, Matsuhisa M (2009) Combination of MafA, PDX-1 and NeuroD is a useful tool to efficiently induce insulin-producing surrogate beta-cells. Curr Med Chem 16:3144–3151
Kim TK, Lee JS, Jung HS, Ha TK, Kim SM, Han N, Lee EJ, Kim TN, Kwon MJ, Lee SH, Kim MK, Rhee BD, Park JH (2014) Triiodothyronine induces proliferation of pancreatic β-cells through the MAPK/ERK pathway. Exp Clin Endocrinol Diabetes 122:240–245
Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU (1985) Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 4:110–125
Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF (2012) Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 61:2205–2213
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P, Karsenty G (2007) Endocrine regulation of energy metabolism by the skeleton. Cell 130:456–469
Lenzen S, Bailey CJ (1984) Thyroid hormones, gonadal and adrenocortical steroids and the function of the islets of Langerhans. Endocr Rev 5:411–434
Ligon B, Ligon B, Yang J, Morin SB, Ruberti MF, Steer ML (2007) Regulation of pancreatic islet cell survival and replication by gamma-aminobutyric acid. Diabetologia 50:764–773
López-Acosta JF, Moreno-Amador JL, Jiménez-Palomares M, Díaz-Marrero AR, Cueto M, Perdomo G, Cózar-Castellano I (2013) Epoxypukalide induces proliferation and protects against cytokine-mediated apoptosis in primary cultures of pancreatic β-cells. PLoS One 8:e52862
Madsbad S, Laurberg P, Weeke J, Orskov H, Faber OK, Binder C, Krarup T, Regeur L (1981) Very early changes in circulating T3 and rT3 during development of metabolic derangement in diabetic patients. Acta Med Scand 209:385–387
Meier JJ, Lin JC, Butler AE, Galasso R, Martinez DS, Butler PC (2006) Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes. Diabetologia 49:1838–1844
Mizokami A, Yasutake Y, Gao J, Matsuda M, Takahashi I, Takeuchi H, Hirata M (2013) Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PLoS One 8:e57375
Moon H, Chon J, Joo J, Kim D, In J, Lee H, Park J, Choi J (2013) FTY720 preserved islet β-cell mass by inhibiting apoptosis and increasing survival of β-cells in db/db mice. Diabetes Metab Res Rev 29:19–24
Nir T, Melton DA, Dor Y (2007) Recovery from diabetes in mice by beta cell regeneration. J Clin Invest 117:2553–2561
Oh YS, Lee YJ, Park K, Choi HH, Yoo S, Jun HS (2014) Treatment with glucokinase activator, YH-GKA, increases cell proliferation and decreases glucotoxic apoptosis in INS-1 cells. Eur J Pharm Sci 51:137–145
Orime K, Shirakawa J, Togashi Y, Tajima K, Inoue H, Ito Y, Sato K, Nakamura A, Aoki K, Goshima Y, Terauchi Y (2013) Trefoil factor 2 promotes cell proliferation in pancreatic β-cells through CXCR-4-mediated ERK1/2 phosphorylation. Endocrinology 154:54–64
Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, Jackson HC, Procter MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C (2006) Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab 3:167–175
Park K (2012) Identification of YH-GKA, a novel benzamideglucokinase activator as therapeutic candidate for type 2 diabetes mellitus. Arch Pharm Res 35:2029–2033
Peshavaria M, Larmie BL, Lausier J, Satish B, Habibovic A, Roskens V, Larock K, Everill B, Leahy JL, Jetton TL (2006) Regulation of pancreatic beta-cell regeneration in the normoglycemic 60 % partial-pancreatectomy mouse. Diabetes 55:3289–3298
Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC (2008) Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10(Suppl 4):32–42
Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC (2013) β-Cell mass and turnover in humans: effects of obesity and aging. Diabetes Care 36:e112
Salpeter SJ, Klein AM, Huangfu D, Grimsby J, Dor Y (2010) Glucose and aging control the quiescence period that follows pancreatic beta cell replication. Development 137:3205–3213
Samson MH, Vestergaard EM, Milman N, Poulsen SS, Nexo E (2008) Circulating serum trefoil factors increase dramatically during pregnancy. Scand J Clin Lab Invest 68:369–374
Samson MH, Poulsen SS, Obeid R, Herrmann W, Nexo E (2011) Trefoil factor family peptides in the human foetus and at birth. Eur J Clin Invest 41:785–792
Sanchis-Gomar F, Perez-Quilis C (2014) The p38-PGC-1α-irisin-betatrophin axis: exploring new pathways in insulin resistance. Adipocyte 3:67–68
Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, Herold KC (2006) Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes 55:3238–3245
Shirakawa J, Okuyama T, Yoshida E, Shimizu M, Horigome Y, Tuno T, Hayasaka M, Abe S, Fuse M, Togashi Y, Terauchi Y (2014) Effects of the antitumor drug OSI-906, a dual inhibitor of IGF-1 receptor and insulin receptor, on the glycemic control, β-cell functions, and β-cell proliferation in male mice. Endocrinology 155:2102–2111
Soltani N, Qiu H, Aleksic M, Glinka Y, Zhao F, Liu R, Li Y, Zhang N, Chakrabarti R, Ng T, Jin T, Zhang H, Lu WY, Feng ZP, Prud’homme GJ, Wang Q (2011) GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc Natl Acad Sci USA 108:11692–11697
Song M, Park HJ (2014) Anti-inflammatory effect of Phellinuslinteus grown on germinated brown rice on dextran sodium sulfate-induced acute colitis in mice and LPS-activated macrophages. J Ethnopharmacol 154:311–318
Sorenson RL, Brelje TC (1997) Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res 29:301–307
Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y (2012) Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem 287:27407–27414
Tian J, Dang H, Chen Z, Guan A, Jin Y, Atkinson MA, Kaufman DL (2013) γ-Aminobutyric acid regulates both the survival and replication of human β-cells. Diabetes 62:3760–3765
Toselli C, Hyslop CM, Hughes M, Natale DR, Santamaria P, Huang CT (2014) Contribution of a non-β-cell source to β-cell mass during pregnancy. PLoS One 9:e100398
Truong W, Emamaullee JA, Merani S, Anderson CC, James Shapiro AM (2007) Human islet function is not impaired by the sphingosine-1-phosphate receptor modulator FTY720. Am J Transpl 7:2031–2038
Vetere A, Choudhary A, Burns SM, Wagner BK (2014) Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov 13:278–289
Wang Y, Liu Y, Wang H, Li C, Qi P, Bao J (2012) Agaricusbisporuslectins mediates islet β-cell proliferation through regulation of cell cycle proteins. Exp Biol Med 237:287–296
Wei P, Shi M, Barnum S, Cho H, Carlson T, Fraser JD (2009) Effects of glucokinase activators GKA50 and LY2121260 on proliferation and apoptosis in pancreatic INS-1 beta cells. Diabetologia 52:2142–2150
Wei J, Hanna T, Suda N, Karsenty G, Ducy P (2014) Osteocalcin promotes β-cell proliferation during development and adulthood through Gprc6a. Diabetes 63:1021–1031
Xiao X, Chen Z, Shiota C, Prasadan K, Guo P, El-Gohary Y, Paredes J, Welsh C, Wiersch J, Gittes GK (2013a) No evidence for β cell neogenesis in murine adult pancreas. J Clin Invest 123:2207–2217
Xiao X, Wiersch J, El-Gohary Y, Guo P, Prasadan K, Paredes J, Welsh C, Shiota C, Gittes GK (2013b) TGFβ receptor signaling is essential for inflammation-induced but not β-cell workload-induced β-cell proliferation. Diabetes 62:1217–1226
Ximenes HM, Lortz S, Jörns A, Lenzen S (2007) Triiodothyronine (T3)-mediated toxicity and induction of apoptosis in insulin-producing INS-1 cells. Life Sci 80:2045–2050
Xu Y, Wang L, He J, Bi Y, Li M, Wang T, Wang L, Jiang Y, Dai M, Lu J, Xu M, Li Y, Hu N, Li J, Mi S, Chen CS, Li G, Mu Y, Zhao J, Kong L, Chen J, Lai S, Wang W, Zhao W, Ning G, 2010 China noncommunicable disease surveillance group (2013) Prevalence and control of diabetes in Chinese adults. JAMA 310:948–959
Yi P, Park JS, Melton DA (2013) Betatrophin: a hormone that controls pancreatic β cell proliferation. Cell 153:747–758
Yi P, Park JS, Melton DA (2014) Perspectives on the activities of ANGPTL8/Betatrophin. Cell 159:467–468
Yu LG, Fernig DG, Smith JA, Milton JD, Rhodes JM (1993) Reversible inhibition of proliferation of epithelial cell lines by Agaricus bisporus (edible mushroom) lectin. Cancer Res 53:4627–4632
Yu LG, Fernig DG, White MR, Spiller DG, Appleton P, Evans RC, Grierson I, Smith JA, Davies H, Gerasimenko OV, Petersen OH, Milton JD, Rhodes JM (1999) Edible mushroom (Agaricus bisporus) lectin, which reversibly inhibits epithelial cell proliferation, blocks nuclear localization sequence-dependent nuclear protein import. J Biol Chem 274:4890–4899
Zhang Y, Li R, Meng Y, Li S, Donelan W, Zhao Y, Qi L, Zhang M, Wang X, Cui T, Yang LJ, Tang D (2014) Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 63:514–525
Zhao Z, Choi J, Zhao C, Ma ZA (2012) FTY720 normalizes hyperglycemia by stimulating β-cell in vivo regeneration in db/db mice through regulation of cyclin D3 and p57 (KIP2). J Biol Chem 287:5562–5573
ACKNOWLEDGEMENTS
We would like to thank Yurika Matsui for comments on this manuscript.
COMPLIANCE WITH ETHICS GUIDELINES
Qiu Li and Zhi-Chun Lai declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Li, Q., Lai, ZC. Recent progress in studies of factors that elicit pancreatic β-cell expansion. Protein Cell 6, 81–87 (2015). https://doi.org/10.1007/s13238-014-0123-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-014-0123-3