
Abstract
Purpose
Metastatic spread of prostate cancer to the skeleton may result in debilitating bone pain. In this review, we address mechanisms underpinning the pathobiology of metastatic prostate cancer induced bone pain (PCIBP) that include sensitization and sprouting of primary afferent sensory nerve fibres in bone. We also review current treatments and pain responses evoked by various treatment modalities in clinical trials in this patient population.
Methods
We reviewed the literature using PubMed to identify research on the pathobiology of PCIBP. Additionally, we reviewed clinical trials of various treatment modalities in patients with PCIBP with pain response outcomes published in the past 7 years.
Results
Recent clinical trials show that radionuclides, given either alone or in combination with chemotherapy, evoked favourable pain responses in many patients and a single fraction of local external beam radiation therapy was as effective as multiple fractions. However, treatment with chemotherapy, small molecule inhibitors and/or immunotherapy agents, produced variable pain responses but pain response was the primary endpoint in only one of these trials. Additionally, there were no published trials of potentially novel analgesic agents in patients with PCIBP.
Conclusion
There is a knowledge gap for clinical trials of chemotherapy, small molecule inhibitors and/or immunotherapy in patients with PCIBP where pain response is the primary endpoint. Also, there are no novel analgesic agents on the horizon for the relief of PCIBP and this is an area of large unmet medical need that warrants concerted research attention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
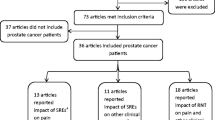
For patients with prostate cancer, bone is the most common organ of metastasis with a prevalence of ~ 90% for men with metastatic castrate-resistant disease [19, 80, 99] and a median survival time of 12–53 months [82]. Metastatic spread of prostate cancer to the skeleton is most often located in the vertebrae (69%) followed by the pelvic bones (41%), long bones (25%) and the skull (14%) [106]. These bone metastases may lead to debilitating bone pain and skeletal-related events (SREs) that not only impair patients’ quality of life (QoL) [4, 82] but are also associated with high healthcare costs and a large socioeconomic burden [82]. SREs include pathological skeletal fracture, spinal cord compression, systemic hypercalcemia and anemia [4]. Of these, spinal cord compression is a medical emergency and may result it permanent paralysis, loss of limb function and mobility, and severe pain [96]. In this review, we address the pathobiology of prostate cancer-induced bone pain (PCIBP), current treatment strategies as well as pain responses from clinical trials published in the past seven years (January 2015 to March 2022) [72].
2 Prostate cancer metastasis to bone
The exact mechanisms underpinning prostate cancer metastasis to bone in preference to other body sites are unclear. One aspect is that the prostate is highly vascularized with the prostatic venous plexus draining into the internal iliac vein, which connects to the vertebral venous plexus throughout the spinal column [9, 80]. The highly vascular trabecular structure of bone provides an ideal environment for metastatic prostate cancer cells to colonize, including ready access to oxygen and other nutrients [60, 80]. The bone marrow is particularly favoured, due to its slower blood flow, high vascularization, and cell composition including osteoclasts and osteoblasts that control bone remodelling [107]. Increased expression of the integrin, αVβ3, on the surface of metastatic prostate tumour cells promotes their adherence to endothelial cells in the bone marrow [93] and expression of receptor activator of nuclear factor kappa-B ligand (RANKL) by prostate cancer cells, promotes their dissemination and colonization of bone [22]. Also, the immunosuppressive tumour microenvironment shields cancer cells from immune surveillance and antitumour activity [4, 80, 86].
In prostate cancer, only rare, phenotypically distinct prostate cancer tumour-initiating cells, also called stem-like prostate cancer cells, have the capacity to form new tumours [59]. However, the mechanisms leading to bone metastasis formation are not fully elucidated [70]. In patients, high expression levels of cyclin A1 and aromatase proteins in metastatic bone lesions support the notion that stem cell-like prostate cancer cells overexpressing cyclin A1 and aromatase, preferentially metastasize to bone [70]. Indeed, cyclin A1 and aromatase increased local production of bone marrow-releasing factors including androgen receptor, Hairy/enhancer-of-split related with YRPW motif-like protein (HeyL), oestrogen, oestrogen receptor alpha, and matrix metalloproteinase 9 (MMP9), that together facilitate metastatic homing to the bone marrow [51, 70, 90]. Thus, local production of steroid hormones and MMPs in the bone marrow likely contribute to generation of a microenvironment suitable for stem-like prostate cancer cells to establish metastatic lesions in bone [51, 70]. The bone microenvironment may also regulate metastatic prostate cancer cells between dormant and proliferative states with dormancy potentially lasting for many months if not years [80, 106].
2.1 Pathobiological bone remodelling in metastatic prostate cancer
Skeletal bone remodelling requires a tight balance between osteoclast-mediated bone resorption and osteoblast mediated bone formation and dysregulation of this balance leads to pathology [38]. At the sites of bone metastases, tumour cells attack the bone and they interact with osteoclasts, osteoblasts, stromal cells and inflammatory cells [109]. Although prostate cancer-induced bone metastasis is primarily osteoblastic in nature, dysregulation of osteoclasts also causes osteolytic lesions [4], which together form osteosclerotic lesions. Prostate tumour cells hyper-secrete endothelin-1 (ET-1), that regulates osteoblast function and contributes potently to PCIBP [4]. Although ET-1 signalling via the endothelin A receptor, stimulates osteoblast proliferation and formation of new bone, this bone is weak and prone to fracture [72, 106]. Activated osteoblasts release RANKL (receptor activator of nuclear factor κB ligand) that interacts with its cognate receptor, RANK, to stimulate proliferation and maturation of osteoclasts which triggers their damaging osteolytic effect on bones [80, 106]. This knowledge led to the development of denosumab (human monoclonal antibody against RANKL) to decrease osteoclastic activity [82]. Although denosumab and bisphosphonates reduce the painful complications of bone metastases, they do not improve overall survival [33].
Osteoclasts produce acidosis-causing protons and adenosine-triphosphate which activate acid-sensing ion channel 3 and the P2 purinoceptor 3 respectively, that are expressed on sensory nerve fibres in bones [103]. Osteoclasts secrete collagenases and proteases that demineralize bone and damage bone matrix protein [106]. Osteoclasts also secrete transforming growth factor-beta and insulin-like growth factor that promote tumour cell proliferation by inhibiting apoptosis [106]. The net outcome is a ‘vicious cycle’ that promotes growth of bone metastases and development of metastatic PCIBP [10].
Within the tumour microenvironment, prostate cancer cells and infiltrating immune cells release an array of pro-inflammatory cytokines including interleukin-1β (IL-1β), IL-6, and tumour necrosis factor alpha (TNFα), as well as chemokines (C–C motif ligands) such as CXCL1, CXCL8 (IL-8), CXCL12 and CCL2, that have a pivotal role in the initiation, progression and metastasis of prostate cancer [58, 71, 100]. However, clinical studies that used monoclonal antibodies to inhibit their pro-tumorigenic effects have been disappointing. For example, addition of the anti-IL-6 monoclonal Ab (mAb), siltuximab (CNT0238) to mitoxantrone/prednisone, reduced expression of IL-6-regulated genes and suppressed known IL-6 signalling pathways including JAK-STAT3 and ERK1/2, but there was no improvement in outcomes for patients with mCRPC [34]. Despite promising efficacy of a CCL2 mAb in rodent models of prostate cancer, sustained suppression of serum CCL2 concentrations was not achieved in patients with mCRPC [83]. A randomized phase Ib/II study (MAGIC-8; NCT03689699) evaluating the effects of nivolumab with or without the IL-8 mAb (BMS-986253), which signals via the chemokine receptors, CXCR1 and CXCR2 [100], in combination with a short course of androgen deprivation therapy in men with castration-sensitive prostate cancer, is currently under investigation. Other ongoing early phase clinical trials in men with mCRPC include the phase 1/2 ACE trial of the small molecule slowly reversible inhibitor of CXCR2, AZD5069, to assess whether its addition to enzalutamide (ADT) will reverse resistance to enzalutamide alone (NCT03177187) [100]. However, blockade of IL-8 signalling via CXCR2 may be insufficient as IL-8 will continue to signal via CXCR1 that is also expressed on prostate cancer cells, stromal and immune cells within the mCRPC microenvironment [100]. To overcome this potential issue, the orally bioavailable small molecule, SX-682, has been developed as a potent allosteric inhibitor of both CXCR1 and CXCR2 [100]. SX-682 is in clinical trials as an add-on treatment to pembrolizumab in patients with metastatic melanoma (NCT03161431), but it may also have applicability in patients with mCRPC [100].
2.2 Sensory and autonomic nerve fibres in bone
Primary afferent sensory nerve fibres and sympathetic nerve fibres are abundant in the three major bone compartments (periosteum, marrow, mineralized bone). The layer of connective tissue that envelops the bone (i.e., periosteum), provides a supportive microenvironment for vasculature, nerves, and periosteal cells [53]. For sensory nerve fibres, the periosteum is the most densely innervated, with fewer sensory fibres in the mineralized bone and the bone marrow (Fig. 1; Table 1) [4, 15, 106].

Primary afferent sensory nerve fibres primarily in the periosteum but also in the bone marrow of prostate cancer invaded bone undergo pathobiological sprouting and sensitization with pronociceptive signalling transduced into the dorsal horn of the spinal cord, from where it projects to the brain where it may be interpreted as pain (Created with Biorender.com)
When expressed by fibre number normalized to tissue area in the distal femur of the mouse, sensory axon innervation in the periosteum was dense (Table 1) with a 15- to 25-fold lower abundance in mineralized bone and the bone marrow (Table 1) [15]. In the latter, unmyelinated calcitonin-gene-related-peptide (CGRP) positive sensory afferents were branched and projected to linear, varicose-rich endings, particularly near the epiphyseal trabecular bone, and less frequently in the metaphysis and diaphysis [15]. Myelinated neurofilament 200-positive sensory axons that mostly expressed CGRP, had a similar pattern of distribution but with a relatively longer and more linear morphology [15]. In bone, there are abundant sympathetic adrenergic neurons that express tyrosine hydroxylase (TH) together with cholinergic neurones containing acetylcholine and vasoactive intestinal peptide [15]. In the bone marrow, larger-diameter blood vessels are often enveloped by sympathetic TH-positive fibres rich in varicosities, and sympathetic axons (like sensory fibres) can dissociate from vasculature to terminate as free-nerve endings in the marrow [15]. Sympathetic nerve fibre density was greatest in the periosteum but less (40%) than that for primary afferent sensory nerve fibres (Table 1) [15]. However, the density of sympathetic fibres in mineralized bone and the bone marrow was considerably higher than that of sensory nerve fibres in the corresponding bone compartments (Table 1) [15].
2.3 Pathobiological bone remodelling and sensitization of nociceptors
The pathobiology of PCIBP is underpinned by neuroplastic changes at multiple levels of the somatosensory system and involves complex cascades of interactions between the metastatic tumour cells, host immune cells, and stromal and tumour-associated factors in the bone microenvironment [108]. The infiltration and proliferation of tumour cells in bone alters the phenotype and function of bone-forming osteoblasts and bone-resorbing osteoclasts, resulting in pathological bone remodelling [15]. This further modulates the expression of various osteoclastogenic and osteoblastogenic factors, such as RANKL, prostaglandin E2 (PGE2), parathyroid hormone-related protein (PTHrP), cytokines (e.g., IL-1, IL-6, IL-8, IL-11, TNF-α), macrophage inflammatory protein 1 α (MIP-1α), endothelin (ET)-1, insulin growth factor 1 (IGF-1), wingless-type protein (Wnt), but to name a few [15, 72, 106, 108]. Of interest, many of these factors are pro-inflammatory mediators and well-known to directly sensitize primary sensory afferent fibres (nociceptors) [15, 55, 72].
The periosteum and bone marrow of an adult bone are uniquely innervated largely by tropomyosin kinase A (TrkA+) thinly myelinated Aδ-fibres and TrkA+ unmyelinated C-fibres, and receive very little innervation by large diameter Aβ fibres and TrkA− C-fibres [49, 55]. These primary afferent fibres express an array of receptors and ion channels, including endothelin A receptors, prostaglandin, TrkA, bradykinin, cytokine, chemokine and purinergic receptors, the transient receptor potential channel, vanilloid subfamily member 1 (TRPV1), and acid-sensing ion channel 3 (ASIC3) (Fig. 1;[4, 44, 55]. The activation of these targets by their cognate ligands from the “pro-inflammatory” soup comprising mediators released from bone stromal cells, osteoclast- and tumour-induced acidosis, and tumour-associated immune cells results in sensitization of nociceptors and transmission of noxious signals to other components of the somatosensory nervous system [55, 72, 103] The intrinsic molecular mechanisms of nociceptor activation by the aforementioned factors are reviewed in detail elsewhere [103, 108].
In addition to sensitization of nociceptors (inflammatory component), seminal works by Mantyh and colleagues have implicated a neuropathic component in the pathobiology of PCIBP. Specifically, studies have shown active and pathological sprouting and neuroma formation by sensory and sympathetic nerve fibres in the periosteum and bone marrow of the metastatic tumour-invaded bone in rodent models of bone cancer pain [14, 49]. This pathological sprouting of nerve fibres was attributed to the actions of nerve growth factor (NGF), and sustained inhibition of NGF, either via administration of anti-NGF or a Pan-Trk inhibitor, markedly attenuated nerve sprouting, neuroma formation and bone pain even in advanced stages of bone cancer [36, 57]. Additionally, administration of anti-NGF (mAb911) has also been shown to suppress functional connectivity alterations in a rodent model of bone cancer pain [20]. However, findings from randomized-controlled clinical trials revealed significant adverse effects of the humanized monoclonal NGF-antibody (tanezumab), including joint damage and rapidly progressive osteoarthritis, resulting in imposition of clinical hold by the Food and Drug Administration on all NGF inhibitors [1].
The ongoing barrage of pronociceptive input mediated by sensory nerve fibres in prostate cancer-invaded bone induces neuroplastic changes (sensitization) in the cell bodies located in the dorsal root ganglia [102]. Augmented release of pronociceptive neurotransmitters at the central terminals in the dorsal horn of the spinal cord, induces multiple changes in the spinal cord to induce a state of central sensitization [98]. This is important as the dorsal horn is a key centre for relaying pronociceptive information from the cancer-invaded bones to 2nd order neurons that project from the spinal cord to higher centres in the brain where this information may be interpreted as pain (Fig. 2) [108].

(Adapted from Zajaczkowska et al. 2019 and redrawn with Biorender.com)
Targets (receptors and ion channels) activated by pronociceptive mediators in metastatic tumour-invaded bone
2.4 Clinical characteristics of bone pain in patients with metastatic prostate cancer
In prostate cancer, the osteosclerotic nature of bone metastases results in the formation of poor-quality bones and as a result SREs are very common [24]. SREs may cause pain, impair physical activity and negatively impact patients’ QoL [99]. Although the best approach for treating metastatic PCIBP is to improve control of the skeletal disease burden [79], this is not always possible. Also, some patients with a single lesion report severe pain whereas others with disseminated bone lesions experience low to moderate pain [103, 106]. Hence, patients need individualized analgesic/ adjuvant medication treatment regimens for alleviation of PCIBP. Of men who die from prostate cancer, > 90% have bone metastases present [29, 84]
PCIBP presents as intermittent dull aches initially, but as metastatic disease progresses, the bone pain becomes constant and more severe with greater intensity during movement and increased severity at night that is not necessarily relieved by lying down [54, 106]. Pain upon palpation is often present in the vicinity of metastatic bone lesions [106]. Ongoing tumour growth within the bone usually leads to breakthrough (episodic) pain, defined as recurrent episodes of extreme pain breaking through the analgesic dosing regimen used to alleviate the background pain [65, 68] Breakthrough pain has a prevalence of ~ 75% in patients with bone metastases, and it is typically acute, piercing and very severe [65, 68]. Breakthrough pain may occur spontaneously without an obvious trigger, or it may be induced by movement and body weight-bearing (incidental) [65, 68]. Breakthrough pain is unpredictable with a short time to maximum pain intensity (< 5 min), and its often-severe intensity has a major negative impact on the ability of patients to undertake activities of daily living and on their QoL [27, 68].
2.5 Summary
Metastasis of prostate cancer to bone dysregulates the normal balance between osteoclasts and osteoblasts in healthy bone remodelling. The net effect is formation of osteosclerotic lesions comprised of poor-quality bone prone to fracture. Additionally, primary afferent sensory nerve fibres innervating the periosteum and the bone marrow of cancer-invaded bones undergo ectopic sprouting. These fibers are then sensitized by a broad array of pronociceptive mediators present in the inflammatory milieu of the cancer-invaded bone. These mediators interact with their cognate receptors and ion channels to transduce pronociceptive signalling into the dorsal horn of the spinal cord and from there to higher centres in the brain where it may be interpreted as pain. PCIBP initially presents as dull aches but as disease progresses, the bone pain becomes constant and more severe in intensity with it worst upon movement and at night. Patients may also experience rapid-onset breakthrough pain that may be spontaneous or triggered by movement and body weight-bearing. Poorly relieved bone pain has a major negative affect on patient’s QoL. Management of bone pain is addressed in the next section of this review.
3 Clinically used analgesic/adjuvant agents for relief of PCIBP
The management of bone metastases in patients with prostate cancer is challenging in terms of morbidity, debilitating pain, impaired function, and poor QoL [45]. Unfortunately, since 2015, there have been no new analgesic agents approved for the relief of PCIBP and so medications succinctly summarized by the World Health Organisation’s 3-Step Analgesic Ladder ([75];Fig. 3), are the mainstay of analgesic therapy [30]. In brief, analgesics for treating background PCIBP include paracetamol and nonsteroidal anti-inflammatory drugs (e.g. ibuprofen and coxibs) for mild pain, with weak opioids such as tramadol or codeine added when pain is mild to moderate in intensity. Weak opioids are replaced by strong opioid analgesics such as morphine when pain is moderate to severe in intensity [30]. Analgesic agents are given orally around the clock [30, 75]. The subcutaneous (s.c.) route is the first-choice alternate route for patients unable to receive opioids by the oral or transdermal routes [30]. Intravenous (i.v.) dosing is an option for opioid titration when rapid pain control is needed and i.v. infusion should be considered when s.c. dosing is contraindicated [30]. For patients with chronic kidney disease with glomerular filtration rates < 30 mL/min), fentanyl and buprenorphine are the safest opioids [30]. When initiating oral morphine therapy for moderate to severe cancer-related pain, it is recommended that patients be individually dose-titrated with immediate-release formulations given every 4 h plus rescue doses (up to hourly) with the daily titrated dose used to convert to a slow-release morphine formulation. The regular dose of slow-release opioids can be adjusted to include the total amount of rescue morphine [30]. Analgesic adjuvant agents such as tricyclic antidepressants (e.g. amitriptyline at doses ≤ 75 mg/kg), duloxetine, and anticonvulsants (e.g. gabapentin, pregabalin) may be added to alleviate a neuropathic component [30, 75]. There is a lack of evidence to support the routine use of the N-methyl-D-aspartate receptor antagonist, ketamine in cancer-related neuropathic pain [30].

World Health Organisation 3-step analgesic ladder for the management of cancer-related pain [76]. (Created with Biorender.com)
For patients with ‘end of dose failure’, the European Society for Medical Oncology (ESMO) guidelines recommend an increase in the dose of strong opioid unless side-effects develop [30]. If the latter, a switch in the dosing route or rotation of the opioid to an equianalgesic dose of a 2nd opioid may be beneficial, and rescue doses are used to treat breakthrough pain [30]. If pain persists despite escalating doses of a strong opioid, it is important to re-assess the pain and treatment, and consider invasive interventions [30]. For the approximately 10% of patients where their cancer pain is difficult to manage with oral or parenteral analgesic agents, interventional techniques include nerve blocks, neurolytic blocks and intrathecal (i.t.) drug delivery [30]. The i.t. route of opioid administration enables logarithmic dose reductions as the opioid is delivered adjacent to the target opioid receptors in the dorsal horn of spinal cord resulting in fewer systemic side-effects [30]. Life expectancy > 6 months justifies the use of an implantable i.t. pump, but only after a trial using a temporary spinal catheter or bolus dose of opioid and local anaesthetic [30].
3.1 Management of opioid-related adverse effects in patients with cancer-related pain
Opioid analgesics evoke a broad array of opioid-related adverse effects with constipation being particularly problematic as tolerance does not develop to constipation. Thus, laxatives must be routinely prescribed for both the prophylaxis and the management of opioid-induced constipation (OIC). The use of naloxone (in association with oxycodone) or methylnaltrexone for managing OIC may be considered [30]. Similarly, naloxegol may be used to treat OIC [27, 76] Anti-dopaminergic drugs and metoclopramide are recommended for the treatment of opioid-related nausea and vomiting [30]. In the event of opioid-induced respiratory depression, naloxone must be used promptly [30].
3.2 Barriers to treatment with opioids
Barriers to appropriate opioid treatment of moderate to severe cancer-related pain such as metastatic PCIBP include fear and apprehension by patients and clinicians [105]. Widespread reporting by the general media of the ‘opioid crisis’, particularly in the United States, may induce opioid phobia in patients leading them to under-report their pain and worsen their suffering [105]. This has led to increased legislative and regulatory activity aimed at tighter controls with respect to opioid prescribing [74]. An unintended consequence may be hesitancy by clinicians to increase opioid dosages for patients with PCIBP due to concerns on addiction, respiratory depression, or decreased life expectancy [105]. To address this issue, the American Society of Clinical Oncology (ASCO) released a policy statement on opioid therapy aimed at protecting access to opioid treatment for cancer-related pain [74]. In particular, patients with cancer and cancer survivors should not be subject to arbitrary prescription limits that artificially limit access to medically necessary treatment with opioids and patients need education on the safe storage of opioid analgesics [74]. Patients may also experience opioid stigma and so interventions targeted to patients, clinicians, and healthcare systems, are needed to de-stigmatize opioid prescription in patients with pain due to advanced cancer such as metastatic prostate cancer [21].
3.3 Rapid-onset opioid analgesic treatments for breakthrough bone pain
Traditionally, breakthrough cancer pain is defined as pain of peak intensity and short duration that occurs in patients who otherwise have stable and acceptable analgesia provided by analgesics given around the clock [61, 65, 68, 69]. The ESMO guidelines recommend oral immediate-release morphine for the relief of breakthrough pain [30], Additionally, fast-onset strong opioid analgesics such as the rapid-onset fentanyl products, are recommended for treating severe breakthrough pain [48, 68]. Their superiority over oral opioids regarding effectiveness and rapid onset of action has been demonstrated in multiple studies [48, 68]. However, their use is restricted to patients who are already receiving, and are tolerant to, opioid analgesics prescribed for managing their background PCIBP to avoid life-threatening respiratory depression [18, 48]. Rapid-onset fentanyl formulations include oral transmucosal products (Table 2) and intranasal fentanyl sprays [7, 27, 37]. Fentanyl is a 100-fold more potent than morphine, it is 90% unionized at physiological pH, and it has high lipophilicity which makes it ideal for oral transmucosal delivery [27]. However, the differential formulation properties and pharmacokinetics of each oral transmucosal fentanyl product (Table 2) mean that they must not be used interchangeably, and each patient must be dose-titrated to an ‘optimal/ successful’ dose to achieve analgesia [18]. The lowest strength dose of each product (Table 2) is recommended to start dose-titration in patients receiving at least 60 mg of oral morphine equivalents (OME) for background pain, which is equivalent to the 3rd step of the WHO Analgesic Ladder [68].
The mechanism(s) underpinning breakthrough pain have been postulated to differ from those transducing background pain that is adequately controlled with strong opioids [41]. To gain insight on this notion, a female rat model of cancer-induced breakthrough pain induced by unilateral intra-tibial injection of mammary adenocarcinoma cells, was developed [41]. In these animals, movement-associated breakthrough pain induced conditioned place avoidance, despite adequate control of ongoing pain by treatment with morphine, thereby mirroring breakthrough pain in the clinical setting [41]. Observation that movement-induced breakthrough pain was abolished by ablation of isolectin B4-binding sensory afferents, but not those expressing TRPV1, implicated input from IB4-binding primary sensory nerve fibers in mediating breakthrough pain [41]. Hence, future research aimed at identifying novel targets specific to this population of sensory nerve fibers, is encouraged to inform novel analgesic drug discovery.
3.4 Summary
Despite decades of research on chronic pain mechanisms, including those that underpin PCIBP, there are no novel analgesic agents for the treatment of cancer-induced bone pain and there are also none on the horizon, despite identification of multiple receptors and ion channels that appear to modulate PCIBP in rodent models. Hence, the pharmacological management of PCIBP continues to be informed by the WHO’s Analgesic Ladder that was first promulgated in 1986. Stigma, opioid phobia by patients and misconceptions by clinicians all contribute to the widespread under treatment of cancer-induced bone pain and this needs to be addressed by educational interventions.
4 Radiotherapy and bone-targeting agents for relief of metastatic PCIBP
Treatment with external beam radiotherapy, radioisotopes, and/or targeted therapy, in association with analgesics, have important roles in the management of metastatic PCIBP [30]. Radiotherapy and bone-targeting therapy are palliative in nature, aimed at improving the patient’s QoL [80].
4.1 Alpha-emitting radionuclides
The alpha-emitter, radium-233 (Ra-233) was the first targeted alpha therapy approved by the United States (US) Food and Drug Administration (FDA) [50, 62] and the European Medicines Agency (EMA) [84]. Importantly, Ra-233 has a significant survival benefit, both in overall survival (OS) and in the time to the first symptomatic SRE [84, 95]. Specifically, in the ALSYMPCA Phase 3 clinical trial (NCT00699751), Ra-233 had a survival benefit of 3.6 months in patients with mCRPC relative to placebo-treated patients [81]. Ra-233 also delayed the onset of symptomatic skeletal events (SSEs) and improved QoL compared with placebo when added to best standard of care [73, 81]. Ra-233 was efficacious irrespective of docetaxel use [43]. Ra-233 is recommended in the ESMO guideline for use in men with bone-predominant, symptomatic mCRPC without visceral metastases [81]. Regarding mechanism of action, Ra-233 accumulates selectively in bone, particularly in areas of high bone turnover such as the border zones of bone and bone metastases, by forming complexes with hydroxyapatite, the inorganic bone matrix, and inducing double-stranded DNA breaks in tumour cells, osteoblasts and osteoclasts [84, 94]. Ra-233 has a highly localised effect as the alpha particles penetrate to a depth of < 0.1 mm in soft tissues which minimises toxicity to nearby healthy tissues [84]. AEs include nausea, diarrhoea, vomiting and peripheral oedema and the most common (> 10%) haematological abnormalities are anaemia, lymphocytopenia, leukopenia, thrombocytopenia and neutropenia [50].
4.1.1 Ra-223 and pain endpoints
The pain responses in patients with mCRPC and treated with Ra-233 either alone or in combination with other treatments in seven clinical studies published in the past 3-years, are summarized in Table 3.
For three of four studies where Ra-233 was given alone, pain responses were the primary endpoint (Table 3). In the PARABO (NCT02398526) study, ~ 60% of patients had a ≥ 2-point decrease in ‘worst pain’ on the Brief Pain Inventory-Short Form (BPI-SF), irrespective of whether or not they were taking opioids [78]. In a small open-label Phase 2 trial of Ra-233 for pain palliation in patients with mCRPC (Table 3), 31% had a decrease in ‘worst pain’ of ≥ 30% from baseline to week 8, which was sustained until week 12 without escalation of pain medication [63]. Pain responders also had a median 53% decrease in pain interference with both general activity and sleep at week 12 (Table 3) [63].
Comparison of high-dose and extended-dosing schedules of Ra-233 in an open label, randomised trial, relative to standard dosing, for treating mCRPC [92] showed that for ‘worst pain’ on the BPI-SF, the pain improvement rates were 27%, 26% & 37% for standard-, high- and extended-dose arms respectively but for the latter this was offset by a higher rate of intolerable side-effects (Table 3).
In a phase 3 trial (NCT02043678), Ra-223 or placebo was combined with oral abiraterone acetate plus oral prednisone or prednisolone (AAP), in patients with chemotherapy-naïve, progressive, mCRPC with ≥ 2 bone metastases (Table 3) [91]. The median time to opioid use was shorter at 19 months for the AAP plus Ra-233 group, c.f. ~ 23 months for the AAP plus placebo group [91]. The less favourable pain response for the combination treatment group was aligned with worse safety outcomes for the same patients (Table 3) [91]. This trial was unblinded prematurely after more fractures and deaths were observed in the Ra-233 group than the placebo group [91]. Overall, addition of Ra-223 to AAP did not improve SSE-free survival and it was associated with increased frequency of bone fractures (29%) compared with placebo (11%) (Table 3) [91]. These findings led to revision of prescribing recommendations for Ra-223 by the FDA and the EMA [91]. A small prospective Phase 2 trial (NCT02507570) of Ra-223 or placebo combined with oral enzalutamide in patients with mCRPC showed that initial benefit on pain responses was lost by the end of treatment [90]. There were no significant safety signals or AEs to contraindicate the combined use of enzalutamide and Ra-233 [90], in contrast to combined dosing of Ra-233 with AAP (Table 3) [91].
4.1.2 Beta-emitting radionuclides
Advantages of beta-emitters including strontium-89 (89Sr), samarium-153 (153Sm), lexidronam (153Sm-EDTMP), phosphorus-32 (32P) sodium phosphate and rhenium-188 (188Re) are their ability to treat multiple disease sites simultaneously, ease of administration, repeatability and potential integration with other treatments [40]. However, in contrast to the α-emitter, Ra-233, these agents do not extend overall survival in patients with mCRPC [82]. However, in March 2022, this situation changed when the FDA approved lutetium (Lu)-177-prostate specific membrane antigen (PSMA)-617 for the treatment of patients with PSMA-positive mCRPC who had been treated previously with an androgen receptor pathway inhibitor and taxane-based chemotherapy [2]. FDA approval was granted based upon the data from the Phase 3 VISION clinical trial (NCT03511664).
4.1.3 β-emitters and pain endpoints
In the open-label Phase 3 VISION trial, PSMA-positive patients with mCRPC and randomized to receive Lu-177-PSMA-617 plus standard of care, had superior pain responses c.f. patients given standard of care alone (Table 3) [88]. Specifically, the median time to worsening pain of ≥ 30% or ≥ 2-points on the BPI-SF, was longer at 5.9 months for the Lu-177-PSMA-617 group c.f. 2.2 months for the standard of care alone group (Table 3) [88]. In other work, patients with mCRPC were randomized to treatment with 10 cycles of docetaxel plus prednisone every 3 weeks with or without rhenium-188-HDEP treatment after the 3rd and 6th cycles of docetaxel (Table 3). The VAS pain scores decreased to 1 and 2 in the control and Re-188 groups respectively, showing similar benefit on pain outcomes between the two groups (Table 3) [97].
4.2 Bone-targeting agents and pain endpoints
Clinical practice guidelines, including the ESMO-endorsed Cancer Care Ontario guideline or the American Society of Clinical Oncology (ASCO) guideline, recommend starting bone-targeted agents (BTA) such as bisphosphonates (zoledronic acid (ZA), pamidronate) or the RANK inhibitor (denosumab) for treating patients with newly diagnosed mCRPC, irrespective of whether or not they are symptomatic [24, 45, 88, 99]. The aim is to prevent SREs and pain progression, as well as stabilization of patient’s QoL (Table 3) [45]. Clinical trial comparison of 4- versus 12-weekly BTA regimens in patients with bone metastases due to mCRPC, showed equivalence for both efficacy and health-related QoL [23, 42]. Thus, de-escalation of denosumab, ZA and pamidronate treatment from 4- to 12-weekly is a reasonable treatment option in these patients [23]. Since widespread adoption of bisphosphonates and denosumab in the routine care of patients with mCRPC, the incidence of SREs has fallen significantly in non-clinical trial populations [99].
In the TRAPEZE study (NCT01057810) (Table 3), pain progression free intervals did not differ significantly in patients with mCRPC randomized to docetaxel plus ZA, docetaxel plus Sr-89, docetaxel plus ZA plus Sr-89 or docetaxel alone, showing no additional benefit on pain responses c.f. that produced by docetaxel alone [47]. In the PROBone study (NCT02410044) (Table 3), 77% of patients with bone metastases due to mCRPC had bone pain at baseline, 15% of whom rated their pain as ‘heavy’ which equated to a VAS score of 9–10 [45]. Once taking a BTA, patients with ‘heavy’ bone pain improved over the 12 month study duration (Table 3) [45]. Additionally, in a network meta-analysis of 21 clinical studies in men with mCRPC and bone metastases, there was little to no difference in pain responses for bisphosphates c.f. placebo/no treatment, and none of the trials reported pain response results for denosumab (Table 3) [46]. Thus, future trials are needed to provide data on pain responses evoked by denosumab in men with mCRPC.
4.3 Local EBRT and pain endpoints
In a systematic review of 26 randomized trials in patients with painful uncomplicated bone metastases due to mCRPC (Table 3), the analgesic efficacy of a single fraction of EBRT did not differ significantly from that of multiple fractions in patients [87]. Overall, 40% of responders achieved pain relief within 10 days of a single fraction and one third of patients achieved complete pain relief (Table 3) [24].
4.4 Exercise-based treatment and pain endpoints
A pilot trial (CHAMP study; NCT02613273) in men with mCRPC who were receiving androgen deprivation therapy (ADT), showed that aerobic and resistance exercise interventions prescribed by an exercise physiologist and remotely monitored, were well tolerated over a 12 week period (Table 3) [50]. Also, patients reported a median overall bone pain VAS score of zero during the 12 week study (Table 3) [50]. The successful tailoring of an exercise program that avoided patients’ metastatic sites to prevent injury, is encouraging [50].
4.5 Systemic therapy (chemotherapy, small molecule inhibitors and immunotherapy)
In the past 5 years, six studies reported on pain responses in patients with PCIBP who received either chemotherapy, small molecule inhibitors, or immunotherapy. However, pain response was a primary outcome in only one of these studies (Table 3). Disappointingly, in this latter randomized phase 3 trial (COMET-2; NCT01522443), the primary pain endpoint was not met irrespective of whether patients received either the tyrosine kinase inhibitor, cabozantinib plus placebo infusions every 3 weeks plus oral prednisone-matched placebo twice-daily, or mitoxantrone every 3 weeks plus twice-daily prednisone plus a once-daily oral cabozantinib-matched placebo (Table 3) [8].
In a phase 3 trial (NCT01057810), asymptomatic/minimally symptomatic, chemotherapy-naïve patients with mCRPC without visceral metastases, and whose disease had progressed during hormonal treatment, and ADT had been discontinued, were recruited and randomized to receive ipilimumab or placebo every 3 weeks for up to 4 doses (Table 3). However, the number of patients with a pain response was too small to evaluate possible treatment-related differences [11]. In the FIRSTANA phase 3 trial (NCT01308567), patients who had chemotherapy-naïve mCRPC, were randomized to cabazitaxel at 20 or 25 mg/m2 (C20 and C25 respectively) or docetaxel at 75 mg/m2 (D75), plus prednisone once-daily, for a median of 9 treatment cycles [77]. Overall, the median times to pain progression-free survival, the percentage of patients with a pain response and those with pain progression did not differ significantly between the three treatment arms (Table 3) [77]. In other work, patients with mCRPC disease progression within 12-months of having received abiraterone or enzalutamide, were randomized (NCT02485691) to cabazitaxel every 3-weeks with once-daily prednisone and granulocyte colony-stimulating factor, or to the other androgen-signaling-targeted inhibitor once-daily [28]. Encouragingly, more than twice as many patients had a confirmed pain response in the cabazitaxel group (45%) c.f. the androgen-signalling-targeted inhibitor group (19%) (Table 3) [28]. Disappointingly, in the ProCAID phase 2 trial (NCT02121639) in patients with mCRPC who received either the pan-AKT inhibitor, capivasertib, or matched placebo in combination with docetaxel & twice-daily prednisone for up to 10 cycles at 3-week intervals, there were no significant differences in bone pain between the two treatment regimens up to treatment cycle 5 (Table 3) [26]. In the TITAN phase 3 trial (NCT02489310), patients with mCRPC who received continuous ADT, and the anti-androgen, apalutamide, had consistently favourable pain scores and a significantly longer time to deterioration of pain compared with those given ADT and placebo, which is encouraging (Table 2) [3].
In summary, from the foregoing, administration of a radionuclide either alone or in combination with chemotherapy, to patients with mCRPC improved pain responses in many patients (Table 3). Additionally, a systematic review of 26 clinical studies showed that a single fraction of local EBRT was as effective as multiple fractions (Table 3). The improved pain responses achieved in a pilot study in patients with mCRPC prescribed aerobic or resistance exercise an exercise-physiologist (Table 3), warrant follow-up with a larger trial. For patients with PCIBP treated with chemotherapy, small molecule inhibitors and/or immunotherapy agents, there were variable pain responses (Table 3). As pain response was the primary endpoint in only one of these six trials (Table 3), future work addressing this gap in knowledge on pain outcomes, is warranted.
5 Conclusion
In men with mCRPC, the prevalence of bone metastases is 90% and median survival time is 12–53 months. Bone metastases may result in SREs and debilitating bone pain that greatly impair the QoL of patients. Current treatments for alleviating PCIBP include radiotherapy, bone-targeting therapy, and analgesic/adjuvant agents that are mainly palliative in nature, aimed at improving pain relief and patient QoL. Recent clinical trials show that radionuclides, given either alone or in combination with chemotherapy, improved pain responses in many patients and a systematic review showed unequivocally that a single fraction of local EBRT was as effective as multiple fractions. However, treatment with chemotherapy, small molecule inhibitors and/or immunotherapy agents, did not improve pain responses in the majority of trials (Table 3). Disappointingly, there are no novel analgesic agents on the horizon for the relief of PCIBP and this is an area of large unmet medical need that warrants concerted research attention.
References
Administration UFaD. Transcript for the March 25, 2021 Joint meeting of the arthritis advisory committee and the drug safety and risk management advisory committee: us food and drug administration; 2021. Accessed 09 May 2022.
Administration UFaD. FDA approves Pluvicto for metastatic castration-resistant prostate cancer 2022 [https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pluvicto-metastatic-castration-resistant-prostate-cancer. Accessed 05 May 2022.
Agarwal N, McQuarrie K, Bjartell A, Chowdhury A, de Santana P, Gomes AJ, Chung BH, et al. Apalutamide plus androgen deprivation therapy for metastatic castration-sensitive prostate cancer: analysis of pain and fatigue in the phase 3 TITAN study. J Urol. 2021;206(4):914–23.
Andriessen A, Donnelly CR, Ji RR. Reciprocal interactions between osteoclasts and nociceptive sensory neurons in bone cancer pain. Pain Reports. 2021;6(1): e867.
Badrising S, Louhanepessy RD, van der Noort V, Kieffer J, Coenen JLLM, Hamberg P, et al. Integrated analysis of pain, health-related quality of life, and analgesic use in patients with metastatic castration-resistant prostate cancer treated with radium-223. Prostate Cancer Prostatic Dis. 2022;25:248–55.
Badrising SK, Louhanepessy RD, van der Noort V, Coenan JLLM, Hamberg P, Beeker A, et al. A prospective observational registry evaluating clinical outcomes of Radium223 treatment in a nonstudy population. Int J Cancer. 2020;147:1143.
Banala S, Khattab OK, Page VD, Warneke CL, Todd KH, Yeung SCJ. Intranasal fentanyl spray versus intravenous opioids for the treatment of severe pain in patients with cancer in the emergency department setting: a randomized controlled trial. PLoS. 2020;15(7): e0235461.
Basch E, Scholz M, de Bono JS, Vogelzang N, de Souza P, Marx G, et al. Cabozantinib versus mitoxantrone-prednisone in symptomatic metastatic castration-resistant prostate cancer: a randomized phase 3 trial with a primary pain endpoint. Eur Urol. 2019;75(6):929–37.
Batson O. The function of the vertebral veins and their role in the spread of metastases. Clin Orthopaed Relat Res. 1995;312(49):4–9.
Battafarano G, Rossi M, Marampon F, Del Fattore A. Cellular and molecular mediators of bone metastatic lesions. Int J Mol Sci. 2018;19(6):1709.
Beer T, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol. 2017;35(1):40–7.
Bertrand A, Iannessi A, Natale R, Beaumont H, Patriti S, Xiong-Ying J, et al. Focused ultrasound for the treatment of bone metastases: effectiveness and feasibility. J Ther Ultrasound. 2018;6(1):1–9.
Bienz M, Saad F. Management of bone metastases in prostate cancer: a review. Curr Opin Support Palliat Care. 2015;9(3):261–7.
Bloom A, Jimenez-Andrade JM, Taylor RN, Castaneda-Corral G, Kaczmarska MJ, Freeman KT, et al. Breast cancer-induced bone remodeling, skeletal pain, and sprouting of sensory nerve fibers. J Pain. 2011;12(6):698–711.
Brazill J, Beeve AT, Craft CS, Ivanusic JJ, Scheller EL. Nerves in bone: evolving concepts in pain and anabolism. J Bone Miner Res. 2019;34(8):1393–406.
Bredenberg S, Duberg M, Lennernas B, Lennernas H, Pettersson A, Westerberg M, et al. In vitro and in vivo evaluation of a new sublingual tablet system for rapid oromucosal absorption using fentanyl citrate as the active substance. Eur J Pharm Sci. 2003;20(3):327–34.
Brito A, Etchebhere E. Radium-223 as an approved modality for treatment of bone metastases. Semin Nucl Med Semin Nucl med. 2020;50(2):177–92.
Brzakala J, Leppert W. The role of rapid onset fentanyl products in the management of breakthrough pain in cancer patients. Pharmacol Rep. 2019;71(3):438–42.
Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, et al. Metastatic patterns of prostate cancer: an autopsy study of 1589 patients. Hum Pathol. 2000;31(5):578–83.
Buehlmann D, Diletta-Ielacqua G, Xandry J, Rudin M. Prospective administration of anti-nerve growth factor treatment effectively suppresses functional connectivity alterations after cancer-induced bone pain in mice. Pain. 2019;160(1):151–9.
Bulls H, Hamm M, Wasilko R, Cameron FA, Belin S, Goodin BR, et al. Manifestations of opioid stigma in patients with advanced cancer: perspectives from patients and their support providers. JCO Oncol Pract. 2022. https://doi.org/10.1200/OP.22.00251.
Chu G, Zhau HE, Wang R, Rogatko A, Feng X, Zayafoon M, et al. RANK- and c-Met-mediated signal network promotes prostate cancer metastatic colonization. Endocr Relat Cancer. 2014;21(2):311–26.
Clemons M, Ong M, Stober C, Ernst S, Booth C, Canil C, et al. A randomised trial of 4—versus 12-weekly administration of bone-targeted agents in patients with bone metastases from breast or castration-resistant prostate cancer. Eur J Cancer. 2021;142:132–40.
Coleman R, Hadji P, Body JJ, Santini D, Chow E, Terpos E, et al. Bone health in cancer: ESMO clinical practice guidelines. Ann Oncol. 2020;31(12):1650–63.
Corn P, Zhang M, Nogueras-Gonzalez GM, Xiao L, Zurita AJ, Subudhi SK, et al. A phase II study of cabozantinib and androgen ablation in patients with hormone-naïve metastatic prostate cancer. Clin Cancer Res. 2020;26(5):990–9.
Crabb S, Griffiths G, Marwood E, Dunkley D, Downs N, Martin K, et al. Pan-AKT Inhibitor capivasertib with docetaxel and prednisolone in metastatic castration-resistant prostate cancer: a randomized, placebo-controlled phase II trial (ProCAID). J Clin Oncol. 2021;39(3):190–201.
Davies A. Oral trnasmucosal opioids cancer-related breakthrough pain. 3rd ed. Oxford: Oxford University Press; 2019.
de Wit R, de Bono J, Sternberg CN, Fizazi K, Tombal B, Wulfing C, et al. Cabazitaxel versus abiraterone or enzalutamide in metastatic prostate cancer. N Engl J Med. 2019;381(26):2506–18.
Den R, George D, Pieczonka C, McNamara M. Ra-223 treatment for bone metastases in castrate-resistant prostate cancer: Practical management issues for patient selection. Am JClin Oncol. 2019;42(4):399.
Fallon M, Giusti R, Aielli F, Hoskin P, Rolke R, Sharma M, et al. Management of cancer pain in adult patients: ESMO clinical practice guidelines. Ann Oncol. 2018;29:166–91.
Feng X, McDonald JM. Disorders of bone remodeling. Annu Rev Pathol. 2011;6:121–45.
Fine P, Streisand JB. A review of oral transmucosal fentanyl citrate: potent, rapid and noninvasive opioid analgesia. J Palliat Med. 1998;1(1):55–63.
Fizazi K, Carducci M, Smith M, Damiao R, Brown J, Karsh L, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377(9768):813–22.
Fizazi K, De Bono JS, Flechon A, Heidenreich A, Voog E, Davis NB, et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur J Cancer. 2012;48(1):85–93.
Galvão D, Taaffe DR, Spry N, Cormie P, Joseph D, Chambers SK, et al. Exercise preserves physical function in prostate cancer patients with bone metastases. Med Sci Sports Exerc Med Sci Sports Exercise. 2018;50(3):393–9.
Ghilardi J, Freeman KT, Jimenez-Andrade JM, Coughlin KA, Kaczmarska MJ, Castaneda-Corral G, et al. Neuroplasticity of sensory and sympathetic nerve fibers in a mouse model of a painful arthritic joint. Arthritis Rheum. 2012;64(7):2223–32.
Giuliani J, Fiorica F, Sacchetto A, Franceschini G, Vaccari F, Bonetti A. The role of fentanyl in the treatment of breakthrough cancer pain: different biotechnologies, different results and different drug costs. J Oncol Pharm Pract. 2021;27(2):445–7.
Hadjidakis D, Androulakis II. Bone remodeling. Ann N Y Acad Sci. 2006;1092:385–96.
Halabi S, Vogelzang NJ, Kornblith AB, Ou SS, Kantoff PW, et al. Pain predicts overall survival in men with metastatic castration-refractory prostate cancer. J Clin Oncol. 2008;26(15):2544–9.
Handkiewicz-Junak D, Poeppel TD, Bodei L, Aktolun C, Ezziddin S, Giammarile F. EANM guidelines for radionuclide therapy of bone metastases with beta-emitting radionuclides. Eur J Nucl Med Mol Imaging. 2018;45(5):846–59.
Havelin J, Imbert I, Sukhtankar D, Remeniuk B, Pelletier I, Gentry J, et al. Mediation of movement-induced breakthrough cancer pain by IB4-binding nociceptors in rats. J Neurosci. 2017;37(20):5111–22.
Hong B, Ibrahim MFK, Fernandes R, Mazzarello S, Hutton B, Shorr R, et al. De-escalation of bone-targeted agents for metastatic prostate cancer. Curr Oncol. 2016;23(1):77–8.
Hoskin P, Sartor O, O’Sullivan JM, Johannessen DC, Helle SI, Logue J, et al. Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: a prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial. Lancet Oncol. 2014;15(12):1397–406.
Hua B, Gao Y, Kong X, Yang L, Hou W, Bao Y. New insights of nociceptor sensitization in bone cancer pain. Expert Opin Ther Targets. 2015;19(2):227–43.
Jakob A, Zahn MO, Nusch A, Werner T, Schnell R, Frank M, et al. Real-world patient-reported outcomes of breast cancer or prostate cancer patients receiving antiresorptive therapy for bone metastases: final results of the PROBone registry study. J Bone Oncol. 2022;33: 100420.
Jakob Tesfamariam T, Macherey S, Kuhr K, Adams A, Monsef I, et al. Bisphosphonates or RANK-ligand-inhibitors for men with prostate cancer and bone metastases: a network meta-analysis. Cochrane Database of Syst Rev. 2020;12(12):13020.
James N, Pirrie SJ, Pope AM, Barton D, Andronis L, Goranitis I, et al. Clinical outcomes and survival following treatment of metastatic castrate-refractory prostate cancer with docetaxel alone or with strontium-89, zoledronic acid, or both: The TRAPEZE randomized clinical trial. JAMA Oncol. 2016;2(4):493–9.
Janknegt R, van den Beuken M, Schiere S, Uberall M, Knaggs R, Hanley J, et al. Rapid acting fentanyl formulations in breakthrough pain in cancer drug selection by means of the system of objectified judgement analysis. Eur J Hosp Pharm. 2018;25(3):1–18.
Jimenez-Andrade J, Mantyh WG, Bloom AP, Xu H, Ferng AS, Dussor G, et al. A phenotypically restricted set of primary afferent nerve fibers innervate the bone versus skin: therapeutic opportunity for treating skeletal pain. Bonekey Rep BoneKEy Rep. 2010;46:306–13.
Van Kenfield S, Blarigan EL, Panchal N, Bang A, Zhang L, Graff R, et al. Feasibility, safety, and acceptability of a remotely monitored exercise pilot CHAMP a clinical trial of high-intensity aerobic and resistance exercise for metastatic castrate-resistant prostate cancer. Cancer Med. 2021;10(22):8058–70.
Kluetz P, Pierce W, Maher VE, Zhang H, Tang S, Song P, et al. Radium Ra 223 dichloride injection u s food and drug administration drug approval summary. Clin Cancer Res Clin Cancer Res. 2014;20(1):9–14.
Lin Q, Cao J, Du X, Yang K, Shen Y, Wang W, Klocker H, et al. The HeyL-aromatase axis promotes cancer stem cell properties by endogenous estrogen-induced autophagy in castration-resistant prostate cancer. Front Oncol. 2021;11(787953):1–13.
Logan J, Jiang J, Shih YCT, Lei X, Xu Y, Hoffman KE, et al. Trends in radiation for bone metastasis during a period of multiple national quality improvement initiatives. J Oncol Pract. 2019;15(4):e356–68.
Lorenz M, Brazill JM, Beeve AT, Shen I, Scheller EL. A Neuroskeletal atlas: spatial mapping and contextualization of axon subtypes innervating the long bones of C3H and B6 mice. J Bone Miner Res. 2021;36(5):1012–25.
Macedo F, Ladeira K, Pinho F, Saraiva N, Bonito N, Pinto L, et al. Bone metastases an overview. Oncol Rev. 2017;11(1):321.
Mantyh P. Bone cancer pain: causes, consequences, and therapeutic opportunities. Pain. 2013;154(Supplement 1):S54–62.
Mantyh P. Bone cancer pain: from mechanism to therapy. Curr Opin Support Palliat Care. 2015;8(2):83–90.
Mantyh W, Jimenez-Andrade JM, Stake JI, Bloom AP, Kaczmarska MJ, Taylor RN, et al. Blockade of nerve sprouting and neuroma formation markedly attenuates the development of late stage cancer pain. Neuroscience. 2010;171(2):588–98.
Maolake A, Izumi K, Natsagdori A, Iwamoto H, Kadomoto S, Makino T, et al. Tumor necrosis factor-alpha induces prostate cancer cell migration in lymphatic metastasis through CCR7 upregulation. Cancer Sci. 2018;109(5):1524–31.
Marcinkiewicz K, Scotland KB, Boorjian SA, Nilsson EM, Persson JL, Abrahamsson PE, et al. The androgen receptor and stem cell pathways in prostate and bladder cancers (Review). Int J Oncol. 2012;40(1):5–12.
Mazo I, von Andrian UH. Adhesion and homing of blood-borne cells in bone marrow microvessels. J Leukoc Biol. 1999;66(1):25–32.
Mazzotta M, Filetti M, Piras M, Mercadante S, Marchetti P, Giusti R. Patients’ satisfaction with breakthrough cancer pain therapy a secondary analysis of IOPS-MS study. Cancer Manag Res. 2022;24(14):1237–45.
McGann S, Horton ER. Radium-223 dichloride: a novel treatment option for castration-resistant prostate cancer patients with symptomatic bone metastases. Ann Pharmacother. 2015;49(4):469–76.
McHugh D, Tagawa S, Moryl N, Milowsky M, Heller G, Osborne J, Rathkopf D, Basch E, Pandit-Taskar N, Morris MJ. a phase ii nonrandomized open trial assessing pain efficacy with radium-223 in symptomatic metastatic castration-resistant prostate cancer. Clin Genitourin Cancer. 2021;19(5):447–56.
RanaMcKay R, Silver R, BhakKorves RHC, Cheng M, Appukkuttan S, et al. Treatment of metastatic castration resistant prostate cancer with radium-223 a retrospective study at a US tertiary oncology center. Prostate Cancer Prostatic Dis. 2021;24(1):210–9.
Mercadante S, Portenoy RK. Breakthrough cancer pain: twenty-five years of study. Pain. 2016;157(12):2657–63.
Mercadante S, Marchetti P, Cuomo A, Mammucari M, Caracenia A. IOPS MS study group breakthrough pain and its treatment critical review and recommendations of IOPS (italian oncologic pain survey) expert group. Support Care Cancer. 2016;24(2):961–8.
Mercadante S, Marchetti P, Cuomo A, Caraceni A, Mediati RD, Mammucari M, et al. Breakthrough cancer pain: preliminary data of the italian oncologic pain multisetting multicentric survey (IOPS-MS). Adv Ther. 2017;34(1):120–35.
Mercadante S, Caraceni A, Masedu F, Scipioni T, Aielli F. Breakthrough cancer pain in patients receiving low doses of opioids for background pain. Oncologist. 2020;25(2):156–60.
Mercadante S, Maltoni M, Russo D, Adile C, Ferrera P, Rossi R, et al. The prevalence and characteristics of breakthrough cancer pain in patients receiving low doses of opioids for background pain. Cancers. 2021;13(5):1058.
Miftakhova R, Hedblom A, Semenas J, Malm J, Persson JL, Miftakhova R, et al. Cyclin A1 and P450 aromatase promote metastatic homing and growth of stem-like prostate cancer cells in the bone marrow. Cancer Res Cancer Res. 2016;76(8):2453–64.
Mughees M, Kaushal JB, Sharma G, Wajid S, Batra SK, Siddiqui JA. Chemokines and cytokines: axis and allies in prostate cancer pathogenesis. Semin Cancer Biol. 2022;16:S1055–579.
Muralidharan A, Smith MT. Pathobiology and management of prostate cancer-induced bone pain: recent insights and future treatments. Inflammopharmacology. 2013;21(5):339–63.
Nilsson S, Cislo P, Sartor O, Vogelsang NJ, Coleman RE, O’Sullivan JM, et al. Patient-reported quality-of-life analysis of radium-223 dichloride from the phase III ALSYMPCA study. Ann Oncol. 2016;27(5):868–74.
American Society of Clinical Oncology. ASCO Policy statement on opioid therapy: protecting access to treatment for cancer-related pain. Alexandria, VA: ASCO; 2016. https://www.asco.org/sites/new-www.asco.org/files/content-files/advocacy-and-policy/documents/2021-Opioids-UPDATE.pdf. Accessed 5 Aug 2022.
The World Health Organisation TWH. Cancer Pain relief. Geneva, Switzerland; 1986.
Ostan R, Gambino G, Malavasi I, Ronga G, Solipaca M, Spunghi M, et al. Can naloxegol therapy improve quality of life in patients with advanced cancer? Cancers. 2021;13(22):5736.
Oudard S, Fizazi K, Seneglov L, Suggard G, Saad F, Hansen S, et al. Cabazitaxel versus docetaxel As First-line therapy for patients with metastatic castration-resistant prostate cancer: a randomized phase III trialFIRSTANA. J Clin Oncol. 2017;35(28):3189–97.
Palmedo H, Ahmadzadehfar H, Eschmann S, Selkinski I, Straube F, Niesen A, et al. 594P Pain efficacy with radium-223 (Ra-223) in patients (pts) with metastatic castration resistant prostate cancer (mCRPC) in the PARABO observational study. Ann Oncol. 2021;32:S641-S.
Pantano F, Zoccoli A, Iuliani M, Lanzetta G, Vincenzi B, Tonini G, et al. New targets, new drugs for metastatic bone pain: a new philosophy. Expert Opin Emerg Drugs. 2011;16(3):403–5.
Park S, Keller ET, Shiozawa Y. Bone marrow microenvironment as a regulator and therapeutic target for prostate cancer bone metastasis. Calcif Tissue Int. 2018;102(2):152–62.
Parker C, Gillessen S, Heidenreich A, Horwich A, Esmo, Guidelines, Committee. Cancer of the prostate: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26:v69–77.
Patel C, Wadas TJ, Shiozawa Y. Progress in targeted alpha-particle-emitting radiopharmaceuticals as treatments for prostate cancer patients with bone metastases. Molecules. 2021;26(2162):1–18.
Pienta K, Michaels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2, in metastatic castration-resistant prostate cancer. Investi New Drugs. 2013;3:760–8.
Poeppel T, Handkiewicz-Junak D, Andreef M, Bercherer A, Bockisch A, Fricke E, et al. EANM guideline for radionuclide therapy with radium-223 of metastatic castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging. 2018;45(5):824–45.
Pound C, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after psa elevation following radical prostatectomy. JAMA. 1999;281(17):1591–7.
Reinstein Z, Pamarthy S, Sagar V, Costa R, Abdulkadir SA, Giles FJ. Overcoming immunosuppression in bone metastases. Crit Rev Oncol Hematol. 2017;117(114):127.
Rich S, Chow R, Raman S, Zeng KL, Lutz S, Lam H, et al. Update of the systematic review of palliative radiation therapy fractionation for bone metastases. Radiother Oncol. 2018;126(3):547–57.
de Sartor O, Bono J, Chi KN, Fizazi K, Hermann K, Rahbar K, et al. Lutetium-177PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med. 2021;385(12):1091–103.
Shen Y, Cao J, Liang Z, Lin Q, Wang J, Yang X, et al. Estrogen receptor a-NOTCH1 axis enhances basal stem-like cells and epithelial-mesenchymal transition phenotypes in prostate cancer. Cell Commun Signaling. 2019;17(1):50.
Shore N, Schellhammer PF, Tutrone RF, Mariados NF, Harrelson SS. Open label phase II study of enzalutamide with concurrent administration of radium 223 dichloride in patients with castration-resistant prostate cancer. Clin Genitourin Cancer. 2020;18(5):416–22.
Smith M, Parker C, Saad F, Miller K, Tombal B, Ng QS, et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet oncol. 2019;20(3):408–19.
Sternberg C, Saad F, Graff JN, Peer A, Vaishampayan UN, Leung E, et al. A randomised phase II trial of three dosing regimens of radium-223 in patients with bone metastatic castration-resistant prostate cancer. Ann Oncol. 2020;31(2):257–65.
Sun Y, Fang M, Wang J, Cooper CR, Pienta KJ, Taichman RS. Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate. 2007;67(1):61–73.
Suominen M, Fagerlund KM, Rissanen JP, Konkol YM, Morko JP, Peng ZQ, et al. Radium-223 inhibits osseous prostate cancer growth by dual targeting of cancer cells and bone microenvironment in mouse models. Clin Cancer Res. 2017;23(15):4335–46.
Terisse S, Karamouza E, Parker CC, Sartor AO, James ND, Pirrie S, et al. Overall survival in men with bone metastases from castration-resistant prostate cancer treated with bone-targeting radioisotopes a meta-analysis of individual patient data from randomized clinical trials. JAMA Oncol. 2020;6(2):206.
Tsuzuki S, Park SH, Eber MR, Peters CM, Shiozawa Y. Skeletal complications in cancer patients with bone metastases. Int J Urol. 2016;23(10):825–32.
van Dodewaard-de Jong J, de Klrek JMH, Bloemendal HJ, Oprea-Lager DE, Hoekstra OS, van denBerg HP, et al. A randomised, phase II study of repeated rhenium-188-HEDP combined with docetaxel and prednisone versus docetaxel and prednisone alone in castration-resistant prostate cancer (CRPC) metastatic to bone; the taxium II trial. Eur J Nuclear Med Mole Imaging. 2017;44(8):1319–27.
Viswanath O, Urits I, Burns J, Charipova K, Gress K, McNally A, et al. Central neuropathic mechanisms in pain signaling pathways: current evidence and recommendations. Adv Ther. 2020;37(5):1946–59.
von Moos R, Body JJ, Egerdie B, Stopeck A, Brown J, Fallowfield L, et al. Pain and analgesic use associated with skeletal-related events in patients with advanced cancer and bone metastases. Support Care Cancer. 2016;24:1327–37.
Waugh D, McGovern JA, McCusker S. The challenges and emerging opportunities of targeting cytokines and chemokine-driven inflammatory signals in metastatic castrate-resistant prostate cancer. Crit Rev Oncog. 2022;27(1):25–43.
Weng W, Huang, LH, Tseng, NC, Ou, YC. Radium-223 for metastatic, castration-resistant prostate cancer: A retrospective chart review study of real-world use in a tertiary hospital in Taiwan. J Formos Med Assoc 2022; 121(10)1929-37.
Yeh T, Luo IW, Hsieh YL, Tseng TJ, Chiang H. Hsieh, ST peripheral neuropathic pain: from experimental models to potential therapeutic targets in dorsal root ganglion neurons. Cells. 2020;9(12):2725.
Yoneda T, Hiasa M, Nagata Y, Okui T, White FA. Acidic microenvironment and bone pain in cancer-colonized bone. Bonekey Rep. 2015;4:690.
Yoneda T, Hiasa M, Okui T, Hata K. Sensory nerves: a driver of the vicious cycle in bone metastasis? J Bone Oncol. 2021;30:1–11.
Yoong J, Poon P. Principles of cancer pain management: an overview and focus on pharmacological and interventional strategies. Aust J Gen Pract. 2018;47(11):758–62.
Zajączkowska R, Kocot-Kępska M, Leppert W, Wordliczek J. Bone pain in cancer patients mechanisms and current treatmentl. J Mol Sci. 2019;20(23):6047.
Zalucha J, Jung Y, Joseph J, Wang J, Berry JE, Shiozawa Y, et al. The role of osteoclasts in early dissemination of prostate cancer tumor cells. J Cancer Stem Cell Res. 2015;3(1):1–14.
Zheng X, Wu YH, Huang JF, Wu AM. Neurophysiological mechanisms of cancer-induced bone pain. J Adv Res. 2022;35:117–27.
Zheng Y, Zhou H, Dunstan CR, Sutherland RL, Seibel MJ. The role of the bone microenvironment in skeletal metastasis. J Bone Oncol. 2013;2(1):47–57.
Acknowledgements
AM is supported by a grant (#RG 20-06) from the Cancer Council New South Wales, Australia. MTS acknowledges the Queensland Government Smart State Research Facilities Programme for supporting CIPDD (Centre for Integrated Preclinical Drug Development) research infrastructure. CIPDD is also supported by Therapeutic Innovation Australia (TIA). TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program.
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Smith, A.E., Muralidharan, A. & Smith, M.T. Prostate cancer induced bone pain: pathobiology, current treatments and pain responses from recent clinical trials. Discov Onc 13, 108 (2022). https://doi.org/10.1007/s12672-022-00569-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-022-00569-z