
Abstract
Recent clinical and epidemiological evidence shows that hormone replacement therapy (HRT) containing both estrogen and progestin increases the risk of primary and metastatic breast cancer in post-menopausal women while HRT containing only estrogen does not. We and others previously showed that progestins promote the growth of human breast cancer cells in vitro and in vivo. In this study, we sought to determine whether apigenin, a low molecular weight anti-carcinogenic flavonoid, inhibits the growth of aggressive Her2/neu-positive BT-474 xenograft tumors in nude mice exposed to medroxyprogesterone acetate (MPA), the most commonly used progestin in the USA. Our data clearly show that apigenin (50 mg/kg) inhibits progression and development of these xenograft tumors by inducing apoptosis, inhibiting cell proliferation, and reducing expression of Her2/neu. Moreover, apigenin reduced levels of vascular endothelial growth factor (VEGF) without altering blood vessel density, indicating that continued expression of VEGF may be required to promote tumor cell survival and maintain blood flow. While previous studies showed that MPA induces receptor activator of nuclear factor kappa-B ligand (RANKL) expression in rodent mammary gland, MPA reduced levels of RANKL in human tumor xenografts. RANKL levels remained suppressed in the presence of apigenin. Exposure of BT-474 cells to MPA in vitro also resulted in lower levels of RANKL; an effect that was independent of progesterone receptors since it occurred both in the presence and absence of the antiprogestin RU-486. In contrast to our in vivo observations, apigenin protected against MPA-dependent RANKL loss in vitro, suggesting that MPA and apigenin modulate RANKL levels differently in breast cancer cells in vivo and in vitro. These preclinical findings suggest that apigenin has potential as an agent for the treatment of progestin-dependent breast disease.
Similar content being viewed by others

Introduction
Post-menopausal women routinely undergo hormone replacement therapy (HRT) containing estrogen alone or a combination of estrogen and progestin to alleviate the climacteric symptoms of menopause such as hot flashes, osteoporosis, insomnia, mood swings, dementia, and decreased libido [1]. Progestins have been included in HRT to counteract the increased risk of endometrial cancer associated with post-menopausal estrogen supplementation. However, the Women’s Health Initiative and subsequent studies of combination HRT showed recently that breast cancer risk is higher in post-menopausal women undergoing estrogen/progestin-based HRT compared with their post-menopausal counterparts given oral estrogen or placebo [2, 3]. While several studies have shown a negative effect of progestins on proliferation of breast cancer cells in vitro [4], progestins increase breast epithelial cell proliferation in vivo [5, 6]. As a result, the use of combination HRT has become controversial and recent epidemiological studies link the decline in HRT to reduced incidence of ductal carcinoma in situ, particularly hormone-receptor-positive invasive breast cancer [7–10]. Due to the rapid onset of tumor growth in postmenopausal women undergoing combination HRT, we and others have suggested that the progestin component promotes the progression of latent tumor cells or stimulates proliferation of cancer stem cells [11, 12].
A number of mechanisms have been proposed to explain progestin-stimulated breast cancer in post-menopausal women [11–13]. Several studies showed that progestins induce the expression of vascular endothelial growth factor (VEGF), a potent pro-angiogenic factor, in human and rodent breast cancer cells [11, 14]. Another study showed that progestins activate several genes that promote cell transformation, increase cell motility, and increase the rate of cancer metastasis [15]. Recently, Schramek et al. [16] and Gonzalez-Suarez et al. [17] reported that progestin induces receptor activator of nuclear factor kappa-B ligand (RANKL), and that higher RANKL may correlate with higher rates of mammary cancer in mice. Kariagina et al. [18] also reported that in cells co-expressing estrogen and progesterone receptors, estrogen and progesterone increase expression of amphiregulin, and that the two hormones co-operate to stimulate robust proliferation of hormone-dependent mammary cancer. Thus, there is an urgent need to identify compounds which can be used in a clinical context to counteract the tumor-promoting effects of progestins in breast cells.
Apigenin is a low molecular weight flavonoid, common in fruits, vegetables, nuts, and plant-derived beverages, that inhibits the growth of human cancer cells in vitro and in vivo ([19 and references therein]). We previously showed that apigenin prevents and/or delays the appearance of medroxyprogesterone acetate (MPA)-dependent 7, 12-dimethylbenz (a) anthracene (DMBA)-induced tumors in vivo [20]. In the present study, we demonstrate that apigenin inhibits the progression and development of MPA-dependent BT-474 xenograft tumors in nude mice via a mechanism which involves apoptosis of proliferating human breast cancer cells.
Materials and Methods
Animals
Female athymic nu/nu nude mice, 5 to 6 weeks old (20–22 g) were purchased from Harlan Sprague–Dawley, Inc. Mice were housed in a laminar air-flow cabinet under specific pathogen-free conditions. All facilities were approved by the American Association for Accreditation of Laboratory Animal Care in accordance with current federal regulations and standards. All surgical and experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Missouri (Columbia, MO) and were in accordance with procedures outlined in the “Guide for Care and Use of Laboratory Animals” (NIH publication 85-23).
Experimental Protocol
We followed the protocol previously described by Liang et al. [21, 22], proceeding as shown in Fig. 1. Briefly, nude mice were implanted with 17-β-estradiol pellets (1.7 mg/pellet, 60-day release; Innovative Research of America, Sarasota, FL) 48 h prior to subcutaneous inoculation with BT-474 tumor cells into both flanks. Tumors were serially measured every 3 days using a digital caliper and tumor volume calculated using the formula (length × width × height) × π/6 [23]. The mean±SE was then calculated for each experimental group for each time point. As previously reported by Liang et al. [21, 22], in this model tumors regress following an initial burst of growth. Once the majority of tumors began to regress, animals were split into two groups (MPA (n = 6) and MPA + apigenin (n = 6)), both of which were implanted with pellets containing MPA (10 mg/pellet, 60-day release) on day 8 following inoculation with tumor cells. A third group (n = 6) was implanted with placebo pellets.

Experimental protocol and timeline for MPA-accelerated human tumor xenografts with apigenin. Nude mice were inoculated with BT-474 cells 2 days after subcutaneous implantation of pellets containing 17ß-estradiol (1.7 mg/60-day release pellet). On day 8 post-inoculation, animals were implanted with either MPA pellets (25 mg/60-day release) or placebo pellets. On day 25, animals given MPA pellets were split into two groups, which received daily i.p. injections of either apigenin (50 mg/kg) or vehicle until day 45 when the experiment was terminated. Animals were observed daily for toxicity, and tumor volume was measured every 3 days
Apigenin was dissolved in 50 % dimethyl sulfoxide (DMSO), 15 % ethinyl alcohol, and 35 % phosphate-buffered saline. Starting on day 25 after inoculation with tumor cells, apigenin (50 mg/kg) or vehicle were administered daily by intra-peritoneal injection for a total of 21 injections. Animals were sacrificed and tumors were collected 3 h after the last injection. Figure 1 summarizes the entire experimental protocol for this study.
Immunohistochemistry
Tumors were collected immediately after the animals were killed and placed in 4 % paraformaldehyde for immunohistochemical (IHC) staining or frozen in liquid nitrogen for future analysis. Collected tissues were then processed for IHC staining using standard procedures previously described by Liang et al. [21]. Stained sections were assessed and analyzed for expression of the following proteins; VEGF, VEGFR-1, VEGFR-2, progesterone receptor (PR), estrogen receptor (ER)-α, ER-β, Ki-67, CD31, and RANKL. Antibodies and dilutions were as follows: anti-PR antibody (1:50 dilution (A0098); DAKO, Carpinteria, CA), anti-ER-α (1:300 (sc-542); Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-ER-ß (1:25 (MCA1974S); AbD Serotec), anti-VEGF antibody (1:100 (sc-152); Santa Cruz Biotechnology, Inc.), anti-VEGFR-1 antibody (1:50 (Flt-1, H225, sc-9029); Santa Cruz Biotechnology, Inc.), anti-VEGFR-2 antibody (1:50 (Flk-1, ab2349); Abcam, Inc., Cambridge, MA), anti-RANKL antibody (1:100 (sc-7628); Santa Cruz Biotechnology, Inc), anti-CD31 antibody (1:100 (ab28364); Abcam, Inc.), anti-Ki-67 antibody (1:600 (RB1510-P); Fisher Scientific), and anti-Her2/neu antibody (1:250 (sc-284); Santa Cruz Biotechnology, Inc).
Tumor samples were stained immunohistochemically and relative staining intensity was quantified using morphometric software (Fovea Pro 3.0, Reindeer Graphics, Asheville, NC). Four images were recorded at ×20 magnification per given tumor, and threshold image intensity was adjusted for measurement in pixels. To determine microvessel density (MVD), four representative photographs of CD31-labeled sections were taken at ×20 magnification for at least five tumor samples per treatment group. The total number of vessels per field was counted. A vessel was defined as an open lumen with adjacent CD31-positive cells. MVD was then expressed as mean microvessel number per field ± SEM.
Proliferation Index
Ki-67 staining was quantified as previously described by Burcombe et al. [24]. A minimum of five randomly selected ×40 high-power fields were examined, each containing representative sections of the tumor with at least 1,500 total cells. Epithelial cells with nuclear Ki-67 immunoreactivity were counted and the proliferation index determined (i.e., number of Ki-67-positive cells divided by total cells).
TUNEL Staining
Tumor tissues were stained by terminal deoxy-nucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL; in situ cell death detection kit, cat no: 11684817910, Roche) and counterstained with haematoxylin and eosin (H&E). The procedure was performed following instructions provided by the manufacturer and in reference to previous studies [25]. A minimum of three randomly selected high-power fields containing representative sections of each tumor were imaged. At least five tumors were examined per treatment group. TUNEL-positive cells and total cells were counted and percent apoptotic cells calculated.
sRANKL Assay
Human sRANKL levels were measured in supernatant collected from cultured BT-474 cells using an enzyme-linked immunoassay kit (RD193004200R, BioVendor, Czech Republic) with a sensitivity of 0.4 pmol/l, an intra-assay coefficient of variation (CV) of 7 % and an inter-assay CV of 11 %. Supernatant was collected from cultured BT-474 cells treated with vehicle (DMSO), MPA (10 nM) ± either apigenin (100 μM) or RU-486 (1 μM) and apigenin or RU-486 alone. All experiments were performed in triplicate following the manufacturer’s recommended protocol, and each sample was analyzed in duplicate.
Statistical Analysis
Statistical significance was tested with one-way ANOVA using SigmaStat Software version 3.5 (Sigmastat Software Inc., Richmond, CA). The assumption of ANOVA was examined and a nonparametric measure based on ranks was used as needed. Values are reported as mean±SEM. When ANOVA indicated a significant effect (p < 0.05), Student–Newman–Keuls post hoc test was used to compare the means of individual groups. When normality test failed, significance was determined by the Kruskal–Wallis test (one-way ANOVA by ranks) followed by the Dunn test as a post hoc test.
Results
Apigenin Prevents Progression of MPA-dependent BT-474 Xenograft Tumors in Nude Mice
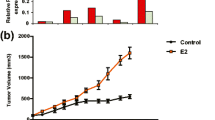
We previously developed a mouse model for studying estrogen- and MPA-dependent xenograft tumors in mice [21, 22]. In the present study, we use this model to examine the effect of apigenin on estrogen and MPA-implanted mice, which develop spontaneous xenograft tumors after subcutaneous inoculation with human BT-474 cells, an aggressive progestin-dependent Her2-positive and p53-defective human breast cancer cell line [21, 22]. The dose regimen and experimental protocol used for this study is shown in Fig. 1. Tumor-bearing animals were injected daily with apigenin (50 mg/kg, i.p.) or vehicle on days 25 to 45 after tumor cell inoculation. As shown in Fig. 2a, tumor size reached a stable plateau rapidly after apigenin dosing began, while tumors grew at a fairly constant rate until animal sacrifice at day 45 in animals dosed with vehicle. The mean tumor size difference reached statistical significance after 12 injections of apigenin. At sacrifice (i.e., day 45), mean tumor size was 208 ± 50 mm3 in animals that were injected with vehicle, 70 ± 10 mm3 in animals injected with apigenin, and 32 ± 7 mm3 in control animals (inoculated with tumor cells but not implanted with an MPA pellet) (Fig. 2a). Representative photographs at the time of animal sacrifice in the three treatment groups are shown in Fig. 2b. Average animal body weight was similar for all three treatment groups (Fig. 2c), and no changes in behavior, eating habits, or mobility were observed in animals treated with apigenin (data not shown).

Apigenin prevents progression of MPA-dependent mammary tumor xenografts in nude mice. a Apigenin inhibited MPA-dependent growth of BT-474 xenograft tumors. b Animals (n = 6 each group) were killed and tumors excised on day 45. Representative tumors from each treatment group are shown. c Graph shows average animal weight per treatment group over entire experimental timeline
As previously noted in the rat model for DMBA-induced MPA-accelerated mammary gland tumors [20], apigenin did not block MPA-induced hyperplasia within tumor-free areas of mammary glands of nude mice (Fig. 3), suggesting that apigenin specifically inhibits proliferation of tumor cells.

Apigenin did not block MPA-induced mammary epithelial hyperplasia in host mice. H&E staining demonstrating the presence of epithelial hyperplasia in animals given MPA alone and MPA + apigenin. Hyperplasia was absent in control animals. Images taken at ×10 magnification
Immunohistochemical Analysis for Her2/neu
BT-474 cells express a high level of Her2/neu, resulting in aberrant growth factor signaling and rapid cell proliferation [26]. Because previous studies showed that apigenin reduces Her2/neu levels in breast cancer cell lines [27], its effect on Her2/neu expression was examined in BT-474 xenograft tumors. As shown in Fig. 4a, apigenin significantly reduced the level of Her2/neu protein in xenograft tumor cells, suggesting that it reduces growth factor signaling via Her2/neu in these cells. Interestingly apigenin did not affect Her2/neu RNA levels, as previously noted [27]. Thus apigenin influences Her2/neu signaling by lowering protein levels, but does not affect Her2/neu gene transcription.

Immunohistochemical analysis of apigenin- and vehicle-treated MPA-induced tumor samples. Representative sections showing a Her2/neu; b TUNEL assay indicating apoptotic cells. TUNEL-positive cells display brownish nuclear staining (red arrows as examples); and c Ki-67. All photographs represent ×40 magnification. Insets represent negative controls without primary antibody. a–c Positive staining was quantified (see “Materials and Methods”), and data are shown as bar graphs. Error bars show standard error of mean. *p < 0.05, significantly different from other treatment groups
Histological and Immunohistochemical Analysis of Markers of Proliferation and Apoptosis
Apigenin could reduce the rate at which a tumor’s size increases by increasing the rate of tumor cell death and/or by decreasing the rate of tumor cell proliferation. To determine which mechanism explains the effect of apigenin on MPA-dependent BT-474 xenograft tumors in nude mice, TUNEL assay was used to measure the fraction of cells undergoing apoptosis in tumors collected from mice either treated with apigenin or not given the flavonoid. We found that apoptotic cells represented a significantly higher fraction (p < 0.05) of tumors from apigenin-treated mice (34 ± 3 %) compared with tumors from untreated (7 ± 1 %) or control mice (14 ± 2 %) (Fig. 4b). Conversely, apigenin slightly lowered immunoreactivity for Ki-67, a marker for cell proliferation, to 47 ± 2 % from 53 ± 1 %, with 45 ± 3 % of control cells staining positively for Ki-67 (Fig. 4c). These data suggest that the anti-tumor effects of apigenin primarily reflect its ability to increase the fraction of tumor cells that undergo apoptosis while it has a smaller effect on tumor cell proliferation.
Immunohistochemical Analysis for PR
Our previous studies showed that the anti-progestin RU-486 blocks the progression and growth of progestin-dependent BT-474 xenograft tumors, suggesting that PR activity is essential for tumor growth [21]. To rule out the possibility that treatment with apigenin leads to a complete loss of PR we assessed PR levels in tumor sections. Our findings concur with previous reports [14, 28], which show that the number of PR-positive cells is significantly lower in mice treated with MPA or both MPA and apigenin (p < 0.05), compared with controls (Fig. 5). Partial loss of PR has been associated with functional PR activity [28, 29] and in this case suggests that apigenin, by virtue of its inability to totally eradicate PR, does not inhibit tumor growth in this model by completely eliminating PR.

Apigenin does not affect MPA-dependent loss of PR in BT-474 tumor xenografts. Upper panel, immunohistochemical staining for PR in representative tumor sections from control and apigenin-treated mice (magnification, ×40). Lower panel, positive staining was quantified (see “Materials and Methods”), and data are shown as a bar graph. Error bars show standard error of mean. *p < 0.05, significantly different from control
Immunohistochemical Analysis of Angiogenic Factors/Blood Vessel Density
In contrast to previous studies from our laboratory [22], the amount of immunoreactive VEGF was similar in tumors from control and MPA-treated animals (Fig. 6a). However, the amount of immunoreactive VEGF was significantly lower in xenograft tumors from apigenin-treated animals. Additional experiments confirmed that levels of expression of VEGF receptor 1 (VEGFR-1 or flt) and VEGF receptor 2 (VEGFR-2 or flk) were similar in animals treated with or without apigenin (data not shown). Based on CD31 immunoreactivity tumors collected from animals in the different treatment groups displayed similar blood vessel densities (Fig. 6b), with average microvessels per field in the range 19 to 22 for all three groups (i.e., control (22 ± 2), MPA (20 ± 2) and MPA + AP (19 ± 1)). However, tumor blood vessel lumen size was smaller in controls and apigenin-treated animals (Fig. 6c), suggesting that the flavonoid might reduce blood flow in BT-474 xenograft tumors.

Immunohistochemical analysis of VEGF and CD-31 in BT-474 xenografts treated with apigenin. Representative tumor samples were analyzed for a VEGF and b CD-31 (magnification, ×40). Positive staining was quantified (see “Materials and Methods”), and data are shown as bar graphs. Error bars show standard error of mean. *p < 0.05, significantly different from control. c Quantification of blood vessel lumen diameter was performed on images with Image-Pro Express 5.0 software. At least ten intra-tumor blood vessels with clear lumen were randomly selected per tissue section and measurements were made for at least four tissues per treatment group. Double asterisks significantly different from control and MPA + AP. *p < 0.05, significantly different from control
Immunohistochemical and ELISA Analysis of RANKL
Recent studies in rodents showed that progestin induces expression of RANKL, and that RANKL may play a significant role in mammary tumor development [16, 17]. Consequently, the effect of apigenin on RANKL expression in xenograft tumor-bearing mice was examined immunohistochemically. In contrast to previous studies in mice, we found that MPA significantly inhibits the production of RANKL in human BT-474 xenograft tumors (p < 0.05), an effect that was independent of co-treatment with apigenin (Fig. 7a). Similarly, when total soluble human RANKL was measured in cultured BT-474 cells, we observed an MPA-dependent lowering of its level, an effect that was not blocked by RU486 but was blocked by 100 μM apigenin (Fig. 7b).

Quantification of RANKL expression in BT-474 xenograft tumors and BT-474 cultured cells. a RANKL expression was measured in xenograft tumors from mice treated with or without apigenin and controls (magnification, ×20). Data are shown as a bar graph. *p < 0.05, indicates significantly different from control. b BT-474 cells in culture were treated with 10 nM MPA ± 1 μM RU or 100 μM AP for 18 h at 37 °C. RANKL was quantified in media from BT-474 cells grown in liquid culture. RU-486 and apigenin were administered 30 min prior to addition of MPA. Soluble RANKL was measured in the supernatant of cultured cells using ELISA (see “Materials and Methods”). Results are expressed as mean±SEM (n = 6). *p < 0.05, significantly different from control
Discussion
In a previous study, we showed that apigenin reduces the frequency of DMBA-induced, progestin-accelerated mammary tumors in rats [20]. The present study examines the effect of apigenin on growth and progression of progestin-dependent BT-474 xenograft tumors in nude mice. BT-474 cells are an aggressive progestin-responsive highly metastatic human breast cancer cell [21, 22]. Under the dose regimen used here (21 daily 50 mg/kg subcutaneous injections of apigenin), apigenin significantly inhibited the growth of BT-474 xenograft tumors. The dose selected was from a previous study [30] and while higher levels of apigenin might prove more potent against tumor growth, this remains to be established. TUNEL assays showed that apigenin induces tumor cell apoptosis, but only modestly inhibits tumor cell proliferation. Wang et al. [31] reported similar findings in colorectal cancer xenografts. Apoptosis is also induced by other tumor growth-inhibiting phytochemicals [32] and anticancer drugs such as 5-flurouracil [33] and cisplatin [34]. Previous studies demonstrating induction of apoptosis by apigenin suggested that the following mechanisms may be involved: induction of caspase 3 [35], induction/activation of p53 [36], up-regulation of FADD [31], and, in MDA-MB-435 cells, stimulation of proteasome-dependent degradation of Her2/neu [27]. In the present study apigenin was associated with reduced tumor cell Her2/neu immunoreactivity, though in agreement with a previous report [27], apigenin did not reduce Her2/neu mRNA levels (not shown). Although it is possible that apigenin stimulates proteasome-dependent degradation of Her2/neu in BT474 xenograft tumors, there are other mechanisms that might alternatively explain reduced immunoreactivity and its consequences. These include reduced Her2/neu autophosphorylation, binding of apigenin to Her2/neu, or interactions that block events downstream of Akt or PI3-kinase. The effect of reduced Her2/neu on tumor cell growth may relate to its potential role in maintaining human breast cancer stem cells [37]. This could be due to the robust regulation of VEGF by Her2/neu [38] which is also associated with maintaining survival of breast tumor cells [39, 40].
As expected, MPA stimulates proliferation of BT-474 xenograft tumor cells, though the proliferation index was modest. This supports the hypothesis that progestins may promote the proliferation of latent breast tumor cells, which may contribute to the increased breast cancer risk associated with progestin-containing HRT [10, 11, 21, 41]. Apigenin blocked MPA-induced proliferation, reducing levels of tumor cell proliferation to those observed in controls. This is consistent with previous studies, and a number of mechanisms by which apigenin could inhibit proliferation have been proposed [19, 42, 43].
In accord with previous studies [14, 28], we observed that MPA inhibits PR expression in human breast cancer cells; in the present study MPA inhibited expression of PR in BT-474 xenograft tumors in both apigenin-treated and untreated animals. As described previously, exogenous progestins stimulate proteasomal degradation of PR [29], which is associated with functional PR activity. Apigenin does not appear to modulate the effects of MPA on PR in tumor cells and thus it is unlikely that it modulates functionality of tumor cell PR, though this remains to be firmly established. In addition, BT-474 cells express androgen receptors (AR) [44], which may also play a role in MPA-dependent tumor growth due to cross-reactivity of the ligand. It is thus possible that some of the inhibitory effects of apigenin occur via modification of AR-mediated growth effects on BT-474 cells, a possibility which remains to be studied.
Contrary to our earlier findings [21, 22], in the present study MPA did not stimulate expression of VEGF in BT-474 xenograft tumors, possibly indicating that VEGF levels are differentially affected by MPA at different stages of tumor growth. However, in this study, basal VEGF immunoreactivity was lower in tumors from apigenin-treated animals. Since VEGF has been shown to be involved in survival and proliferation of mammary tumor cells [39, 40, 45, 46], it is possible that loss of the angiogenic growth factor leads to tumor cell loss. Furthermore, although blood vessel density was similar in tumors from all animal groups, apigenin-treatment correlated with smaller blood vessel lumen size. It is therefore conceivable that apigenin restricts tumor blood supply, though the mechanism through which this occurs remains to be determined.
Recent studies show that MPA induces RANKL in DMBA-induced mammary tumors, which is consistent with a role for RANKL in the growth of progestin-dependent breast cancer [16, 17]. Because MPA stimulates proliferation of BT-474 xenograft tumor cells, we examined whether RANKL might also be induced by MPA in BT-474 cells. However, in contrast to findings in rodents, RANKL expression was lower in BT-474 xenograft tumor cells from animals treated with MPA (with or without apigenin) than in controls (animals not exposed to MPA). A similar effect was seen in vitro; MPA inhibited expression of RANKL in cultured BT-474 cells, an effect that was not blocked by RU-486, but was blocked by apigenin. The significance of this observation remains to be determined. A previous study indicated that membrane-associated PR is insensitive to the negative regulatory effect of antiprogestins [47], thus it is possible that membrane-associated PR mediates MPA-dependent down-regulation of RANKL. Moreover, the effect of progestin-dependent RANKL on human breast cancer progression is controversial [48], and it is possible that progestin regulates expression of RANKL via different mechanisms in murine and human mammary cells [49]. In vitro, progestins generally reduce the proliferation of human breast cancer cells [4]. It is therefore possible that in vitro inhibition of RANKL by progestins is associated with inhibition of progestin-mediated cell proliferation in vitro, a process which appears to occur differently in vivo, possibly due to epithelial-stromal interactions modifying the signal. Earlier studies reported that serum levels of RANKL are unaffected by progestin-containing HRT in postmenopausal women [50]. However, it is possible that different tumors (or different cells within a single tumor) might differentially induce or suppress expression of RANKL in response to MPA. Further studies are needed; in particular, the effect of progestin on expression of RANKL should be evaluated in other human breast cancer cells. It is worth noting that progesterone exerts strain-specific effects on RANKL expression in mice [51].
Although this study shows that apigenin blocks breast cancer xenograft tumor growth by inducing apoptosis, apigenin does not affect proliferation and hyperplasia of ducts and lobular epithelial cells in the normal mammary gland of nude mice. Similar observations were made by Mafuvadze et al. [20] in DMBA-induced progestin-accelerated rat mammary tumors and by Gupta et al. [52], who suggested that apigenin may exert different effects on prostate cancer cells than on normal cells. By virtue of its ability to selectively induce apoptosis in rapidly growing breast cancer cells, while having no effect on normal mammary cells, apigenin would seem to be an extremely promising chemotherapeutic agent.
In conclusion, we show that apigenin inhibits the growth of MPA-dependent BT-474 xenograft tumors, both by inducing apoptosis and by inhibiting proliferation of tumor epithelial cells. Apigenin may also reduce the production by tumor cells of pro-angiogenic VEGF, which is essential for growth and maintenance of blood vessels in the tumor microenvironment. The use of antiprogestins as a therapeutic agent was previously explored [53], and mostly phased out due to toxicities and inconsistent effects on tumor shrinkage. The potential use of a natural antiprogestin would therefore appear to possess a number of advantages. Although the pharmacokinetics and effectiveness of apigenin in humans need to be evaluated, we contend that combination therapy including apigenin and other chemotherapeutic agents warrants further study. For example, the combined use of apigenin and tamoxifen or aromatase inhibitors should be explored, since, as we show in this study, apigenin does not affect in vivo ER expression in breast cancer cells. Future studies should also examine the effects of apigenin on breast cancer progression promoted by other synthetic progestins and the endogenous hormone progesterone.
References
Narod SA (2011) Hormone replacement therapy and the risk of breast cancer. Nat Rev Clin Oncol 8:669–676
Ross RK, Paganini-Hill A, Wan PC, Pike MC (2000) Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J National Cancer Inst 92:328–332
Chlebowski RT, Anderson GL, Gass M et al (2010) Writing group for Women’s Health Initiative Investigators. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 304:1719–1720
Clarke CL, Sutherland RL (1990) Progestin regulation of cellular proliferation. Endocr Rev 11:266–301
Aupperlee M, Kariagina A, Osuch J, Haslam SZ (2005–2006) Progestins and breast cancer. Breast Dis 24:37–57
Hyder SM, Chiappetta C, Stancel GM (2001) Pharmacological and endogenous progestins induce vascular endothelial growth factor expression in human breast cancer cells. Int J Cancer 92:469–473
Tsai S, Stefanick ML, Stafford RS (2011) Trends in menopausal hormone therapy use of US office-based physicians, 2000–2009. Menopause 18:385–392
Ravdin PM, Cronin KA, Howlader N, Berg CD, Chlebowski RT et al (2007) The decrease in breast cancer incidence in 2003 in the United States. N Engl J Med 356:1670–1674
Ereman RE, Prebil LA, Mockus M, Koblick K, Orenstein F, Benz C, Clarke CA (2010) Recent trends in hormone therapy utilization and breast cancer incidence rates in the high incidence population of Marin County, California. BMC Publ Health 10:228
Clarke CA, Glasse SL, Uratsu CS, Selby JV, Kushi LH, Herrinton LJ (2006) Recent declines in hormone therapy utilization and breast cancer incidence: clinical and population-based evidence. J Clin Oncol 24:e49–e50
Hyder SM, Murthry L, Stancel GM (1998) Progestin regulation of vascular endothelial growth factor in human breast cancer cells. Cancer Res 58:392–395
Joshi PA, Jackson HW, Beristain AG et al (2010) Progesterone induces adult mammary stem cell expansion. Nature 465:803–807
Lanari C, Molinolo AA (2002) Progesterone receptors—animal models and cell signaling in breast cancer: diverse activation pathways for the progesterone receptor possible implications for breast biology and cancer. Breast Cancer Res 4:240–243
Benakanakere I, Besch-Williford C, Schnell J, Brandt S, Ellersieck MR, Molinolo A, Hyder SM (2006) Natural and synthetic progestins accelerate 7,12-dimethylbenz[a]anthracene-initiated mammary tumors and increase angiogenesis in Sprague–Dawley rats. Clin Cancer Res 12:4062–4071
McGowan EM, Clarke CL (1999) Effects of overexpression of progesterone receptor A on endogenous progestin-sensitive endpoints in breast cancer cells. Mol Endocrinol 13:1657–1671
Schramek D, Leibbrandt A, Sigl V et al (2010) Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 468:98–102
Gonzalez-Suarez E, Jacob AP, Jones J et al (2010) RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 468:103–107
Kariagina A, Xie J, Leipprandt JR, Haslam SZ (2010) Amphiregulin mediates estrogen, progesterone and EGFR signaling in the normal rat mammary gland and in hormone-dependent rat mammary cancers. Horm Cancer 1:229–244
Patel D, Shukla S, Gupta S (2007) Apigenin and cancer chemoprevention: progress, potential and promise. Int J Oncol 30:233–245
Mafuvadze B, Benakanakere I, Lopez Perez FR, Besch-Williford C, Ellersieck MR, Hyder SM (2011) Apigenin prevents development of medroxyprogesterone acetate-accelerated 7, 12-dimethylbenz (a) anthracene-induced mammary tumors in Sprague–Dawley rats. Cancer Prev Res 4:1316–1324
Liang Y, Besch-Williford C, Brekken RA, Hyder SM (2007) Progestin-dependent progression of human breast tumor xenografts: a novel model for evaluating anti-tumor therapeutics. Cancer Res 67:9929–9936
Liang Y, Benakanakere I, Besch-Williford CB, Hyder RS, Ellersieck M, Hyder SM (2010) Synthetic progestins induce growth and metastasis of BT-474 human breast cancer xenografts in nude mice. Menopause 17:1040–1047
El Etreby MF, Liang Y (1998) Effect of antiprogestins and tamoxifen on growth inhibition of MCF-7 human breast cancer cells in nude mice. Breast Cancer Res Treat 49:109–117
Burcombe R, Wilson GD, Dowsett M, Khan I, Richman PI, Daley F, Detre S, Makris A (2006) Evaluation of Ki-67 proliferation and apoptotic index before, during and after neoadjuvant chemotherapy for primary breast cancer. Breast Cancer Res 8:R31
Sun P, Ren X, Zhang H, Li X, Cai S, Ye K, Li X (2003) Serum from rabbit orally administered cobra venom inhibits growth of implanted hepatocellular carcinoma cells in mice. World J Gastroenterol 9:2441–2444
Saal LH, Holm K, Maurer M, Memeo L, Su T et al (2005) PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res 65:2554–2559
Way T, Kao M, Lin J (2004) Apigenin induces apoptosis through proteasomal degradation of HER2/neu in HER2/neu-overexpressing breast cancer cells via the phosphatidylinositol 3-kinase/akt-dependent pathway. J Biol Chem 279:4479–4489
Dennis AP, Lonard DM, Nawaz Z, O’Malley BW (2005) Inhibition of the 26S proteasome blocks progesterone receptor-dependent transcription through failed recruitment of RNA polymerase II. J Steroid Biochem Mol Biol 94:337–346
Ellmann S, Sicht H, Thiel F et al (2009) Estrogen and progesterone receptors: from molecular structures to clinical targets. Cell Mol Life Sci. doi:10.1007/s00018-009-0017-3
Chen D, Landis-Piwowar KR, Chen MS, Dou QP (2007) Inhibition of proteasome activity by the dietary flavonoid apigenin is associated with growth inhibition in cultured breast cancer cells and xenografts. Breast Cancer Res Treat 9:R80
Wang QR, Yao XQ, Wen G, Fan Q, Li Y, Fu XQ, Li CK, Sun XG (2011) Apigenin suppresses the growth of colorectal cancer xenografts via phosphorylation and up-regulated FADD expression. Oncol Lett 2:43–47
Zhao AG, Zhao HL, Jin XJ, Yang LD (2002) Effects of Chinese jianpi herbs on cell apoptosis and related gene expression in human gastric cancer grafted onto nude mice. World J Gastroenterol 8:792–796
Hsueh C, Kelsen D, Schwartz GK (1998) UCN-01 suppresses thymidylate synthase gene expression and enhances 5-fluorouracil-induced in a sequence dependent manner. Clin Cancer Res 4:2201–2206
Henkels KM, Turchi JJ (1999) Cisplatin-induced apoptosis proceeds by caspase-3 dependent and independent pathways in cisplatin-resistant and sensitive human ovarian cancer cell lines. Cancer Res 59:3077–3083
Vargo MA, Voss OH, Poustka F, Cardounel AJ, Grotewold E, Doseff AI (2006) Apigenin-induced-apoptosis is mediated by the activation of PKCdelta and caspases in leukemia cells. Biochem Pharmacol 72:681–692
Zheng P, Chiang L, Lin C (2005) Apigenin induced apoptosis through p53-dependent pathway in human cervical carcinoma cells. Life Sci 76:1367–1379
Korkaya H, Paulson A, Iovino F, Wicha MS (2008) HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 27:6120–6130
Le X-F, Eiqun Mao W, Lu C, Thornton A, Heymach JV, Sood AK, Bast RC Jr (2008) Specific blockade of VEGF and HER2 pathways results in greater growth inhibition of breast cancer xenografts that overexpress Her2. Cell Cycle 7:3747–3758
Liang Y, Hyder SM (2005) Proliferation of endothelial and tumor epithelial cells by progestin-induced vascular endothelial growth factor from human breast cancer cells: paracrine and autocrine effects. Endocrinology 146:3632–3641
Pidgeon GP, Barr MP, Harmey JH, Foley DA, Bouchier-Hayes DJ (2001) Vascular endothelial growth factor (VEGF) upregulates BCL-2 and inhibits apoptosis in human and murine mammary adenocarcinoma cells. Br J Cancer 85:273–278
Horwitz KB, Sartorius CA (2008) Progestins in hormone replacement therapies reactivate cancer stem cells in women with preexisting breast cancers: a hypothesis. J Clin Endocrinol Metab 93:3295–3298
Ujiki MB, Ding X, Salabat MR, Bentrem DJ, Golkar L, Milam B, Talamonti Bell RH, Iwamura T, Adrian TE (2006) Apigenin inhibits pancreatic cancer cell proliferation through G2/M cell cycle arrest. Mol Cancer 5:76
Zhao M, Ma J, Zhu H-Y, Du Z, Xu Y, Yu X (2011) Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol Cancer 10:104
Magklara A, Brown TJ, Diamandis EP (2002) Characterization of androgen receptor and nuclear receptor co-regulator expression in human breast cancer cell lines exhibiting differential regulation of kallikreins 2 and 3. Int J Cancer 100:507–514
Lee T, Seng S, Sekine M, Hinton C, Fu Y, Avraham HK, Avraham S (2007) Vascular Endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed VEGFR1/FLT1. PLoS Med 4:e186
Su J, Yen C, Chen P, Chuang S, Hong C, Kuo I, Chen H, Hung M, Kuo M (2007) The role of the VEGF-C/VEGFR-3 axis in cancer progression. Br J Cancer 96:541–545
Bottino MC, Cerliani JP, Rojas P, Giulianelli S, Soldati R, Mondillo C et al (2011) Classical membrane progesterone receptors in murine mammary carcinomas: agonistic effects of progestins and RU-486 mediating rapid non-genomic effects. Breast Cancer Res Treat 126:621–636
Petrie WK, Hovey RC (2011) A local basis for progesterone action during mammary tumorigenesis—no longer RANK and file. Breast Cancer Res 13:301
Tanos T, Brisken C (2011) High hopes for RANKL: will the mouse model live up to its promise? Breast Cancer Res 13:302
Di Carlo C, Tommaselli GA, Gargano V, Sammartino A, Bifulco G, Tauchmanova L, Colao A, Nappi C (2007) Effects of estrogen-progestin on serum levels of RANKL, osteoprotegerin, osteocalcin, leptin, and ghrelin in postmenopausal women. Menopause 14(1):38–44
Aupperlee MD, Drolet AA, Durairaj S, Wang W, Schwartz RC, Haslam SZ (2009) Strain-specific differences in the mechanisms of progesterone regulation of murine mammary gland development. Endocrinology 150:1485–1494
Gupta S, Afaq F, Mukhtar H (2001) Selective growth-inhibitory, cell-cycle deregulatory and apoptotic response of apigenin in normal versus human prostate carcinoma cells. Biochem Biophys Res Commun 287:914–920
Klijn JG, de Jong FH, Bakker GH, Lamberts SW, Rodenburg CJ, Alexieva-Figusch J (1989) Antiprogestins, a new form of endocrine therapy for human breast cancer. Cancer Res 49:2851–2856
Acknowledgments
This research was supported by COR funds and research funds from RADIL, University of Missouri, Columbia. SMH is the Zalk Missouri Professor of Tumor Angiogenesis. We wish to thank Dr. F. Lopez for assistance with the photographs shown in the manuscript as well as the RADIL staff for assistance with immunohistochemistry.
Conflict of Interest
The authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mafuvadze, B., Liang, Y., Besch-Williford, C. et al. Apigenin Induces Apoptosis and Blocks Growth of Medroxyprogesterone Acetate-Dependent BT-474 Xenograft Tumors. HORM CANC 3, 160–171 (2012). https://doi.org/10.1007/s12672-012-0114-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-012-0114-x