
Abstract
Acute opioid withdrawal syndrome is a series of neurological symptoms caused by the abrupt cessation of the chronic administration of opioids such as morphine. The locus coeruleus (LC) in the brain stem receives a dense projection of orexinergic fibers from the hypothalamus and is a candidate site for the expression of the somatic aspects of morphine withdrawal. Previous studies have shown that orexin-A contributes to the behavioral symptoms of naloxone-induced morphine withdrawal, partly by reducing the activity of GABAergic neurons, suggesting that orexin-A may negatively modulate fast GABAergic neurotransmission during morphine withdrawal. We used whole-cell patch-clamp recordings of LC neurons in brainstem slices to investigate the effect of orexin-A on bicuculline-sensitive GABAergic inhibitory postsynaptic currents (IPSCs) during naloxone-induced morphine withdrawal. Male Wistar rats (P14–P21) were given morphine (20 mg/kg, i.p.) daily for seven consecutive days to create dependency on the drug. The application of naloxone (1 µM) to brain slices of morphine-treated rats reduced the amplitude of evoked IPSCs (eIPSCs) as well as spontaneous IPSCs (sIPSCs) frequency but did not change sIPSCs amplitude. Orexin-A (100 nM) significantly enhanced the suppressive effect of naloxone on eIPSCs amplitude and sIPSCs frequency but had no effect on the presence of the orexin type 1 receptor (OX1R) antagonist, SB-334867. Orexin-A alone had no significant effect on eIPSCs and sIPSCs in the absence of naloxone. In summary, our results show that orexin-A, via OX1R, potentiates the suppressive effect of naloxone on GABAergic IPSCs of LC neurons in morphine-treated rats. We conclude that orexins may have a critical role in regulating GABAergic neurotransmission to LC neurons during naloxone-induced morphine withdrawal.
Similar content being viewed by others

Introduction
Morphine is an archetypal opioid analgesic that modulates neuronal function via activation of μ-opioid receptors (MORs). Chronic administration of morphine creates dependency on the drug such that abrupt cessation, either by discontinuation of treatment or application of a MOR antagonist, results in an acute opioid withdrawal syndrome. Morphine withdrawal is characterized by multiple neurological symptoms including perspiration, nausea, anxiety, drug cravings, followed by pain hypersensitivity and loss of appetite [1]. Behavioral signs of withdrawal may also be observed in morphine-treated rats upon treatment with the MOR antagonist naloxone [2, 3].
The locus coeruleus (LC) nucleus of the brain stem is a candidate site for the expression of autonomic dysfunction and other symptoms following morphine withdrawal [4]. The LC plays an important role in the regulation of multiple physiological processes including attention, pain, sleep–wake cycle, learning, memory, stress, and addiction [5,6,7,8]. LC neurons are characterized by their spontaneous firing activity and pacemaker activity, which in turn is modulated by a number of neurotransmitters and neuropeptides.
Orexins are hypothalamic neuropeptides that are responsible for regulating arousal and appetite. Anatomical studies report that the lateral hypothalamus sends orexinergic excitatory inputs to LC neurons [9]. There is also evidence that the orexin isoform orexin-A (also known as hypocretin 1) can result in the depolarization of LC neurons and an increase in their firing rate [10, 11]. The stimulatory effect of orexin-A in LC neurons is mediated via high-affinity orexin type 1 receptors (OX1R) [11, 12], which are expressed at high density in the LC nucleus [13].
Several studies have suggested that activation of OX1Rs in the LC nucleus contributes to the development of morphine dependency [14,15,16,17]. For example, the behavioral signs of morphine withdrawal are attenuated in orexin knock-out mice and are reduced by OX1R blockade in rats [18, 19]. More recently, we have demonstrated that microinjection of the OX1R antagonist, SB-334867, into the LC can also attenuate naloxone-induced morphine withdrawal behaviors in rats [20,21,22].
Orexinergic signaling has previously been shown to reduce GABAergic neurotransmission in the brain stem nucleus ambiguus [23] and to decrease GABA release in the ventrolateral periaqueductal gray matter of the rat [24]. Interestingly, injection of the GABAA receptor agonist muscimol, into the LC reduced the behavioral signs of naloxone-induced morphine withdrawal [25] and the effect of OX1R antagonism in the LC could be reversed by pre-treatment with the GABAA-receptor antagonist bicuculline [20].
Together, the above studies suggest that at least some of the behavioral symptoms of opioid withdrawal are due to an OX1R-mediated reduction of inhibitory GABAergic tone within the LC. However, an effect of orexin-A on fast GABAergic synaptic transmission in the LC has not yet been reported. We therefore investigated the effect of orexin-A on evoked and spontaneous IPSCs in LC neurons of brainstem slices from morphine-treated rats, in an in vitro model of naloxone-induced opioid withdrawal.
Materials and methods
Ethics statement
All experiments were conducted in accordance with the ethical guidelines of the Faculty of Medical Sciences, Tarbiat Modares University based on the United States NIH Guide for the Care and Use of Laboratory Animals. Attempts were made to minimize animal suffering and to decrease the number of animals used.
Animals
All electrophysiological recordings were performed in brainstem slices prepared from 2–3-week-old male Wistar rats (Pasteur institute of Tehran, Iran). Rats received water and regular rat chow ad libitum and were housed under climate-controlled conditions with a 12-h light/dark cycle (the light period started at 7 a.m.). In order to diminish animals’ stress, they were gently handled before the experiments. All experiments were carried out during the light phase (12:00–16:00).
Drugs
The main chemicals used in these experiments were morphine sulfate (Temad, Iran), orexin-A and SB-334867 (Tocris, UK), diethyl ether (Merck, Germany); naloxone hydrochloride, bicuculline, AP-5, CNQX, QX-314 and D-Phe-Cys-Tyr-D-Trp-ArgThr-Pen-Thr-NH2 (CTAP) were all purchased from Sigma (Sigma, USA).
Injection of drugs
Morphine and naloxone were freshly dissolved in physiological saline. For induction of morphine dependency, animals were chronically treated with morphine (20 mg/kg, i.p.) once a day for seven consecutive days [26]. On the 8th day, to confirm the morphine dependency, a group of animals was injected with naloxone and then behavioral manifestations of withdrawal (head moves, moving paws, rolling, and stretches) were monitored for 25 min, exactly as described previously [27] (Table 1). The brain slices of morphine-treated rats were continuously bathed in morphine (5 μM) from the beginning to the end of the experiments to prevent spontaneous opioid withdrawal. In addition, naloxone (1 μM) was superfused in slices containing LC neurons to induce morphine withdrawal after 10 min baseline recording.
Whole-cell patch clamp
Seven days after morphine treatment, the rats were anesthetized with diethyl ether and immediately decapitated. The rats’ brains were rapidly extracted from the skull. Subsequently, the brain was trimmed in ice-cold (1–4 °C), low-calcium sucrose-based artificial cerebrospinal fluid (sucrose-aCSF) containing (in mM): sucrose 213, KCl 2.6, CaCl2 0.1, MgCl2 2, NaHCO3 26, NaH2PO4 1.23, l-ascorbic acid 0.4, d-glucose 2 (285–295 mOsmol/l, pH 7.3–7.4 when bubbled with 95% O2, 5% CO2). Horizontal brain slices (250–300 μm thick), containing the LC, were cut on a vibratome (1000 Plus Sectioning System, Vibratome, USA). The slices were transferred to a holding chamber and incubated with standard aCSF containing (in mM): NaCl 125, KCl 3, NaH2PO4 1.25, NaHCO3 25, CaCl2 2, MgCl2 1, l-ascorbic acid 0.4 and d-glucose 10, saturated with 95% O2 and 5% CO2 for 45 min at 35 °C, pH 7.3–7.4 (osmolarity, 295–305 mOsmol/l).
Brain slices were transferred to the recording chamber on a fixed-stage upright microscope (Axioskop 2 FS, Carl Zeiss, Germany) and secured under nylon strings attached to a U-shaped platinum frame. Slices were constantly perfused with standard aCSF at a flow rate of 1–2 ml/min. Neurons were visualized using an infrared CCD camera (IR-1000, USA) with 10× and 40× water immersion objective lenses. Electrodes (resistances, 3–7 MΩ) were made from borosilicate glass capillaries (1.5 mm outer diameter, GC150-11; Harvard Apparatus, UK) with a programmable puller (P-97, Sutter Instrument, USA). Patch pipettes were filled with intracellular solution containing (in mM): CsCl 140, NaCl 6, CaCl2 1, MgCl2 2, MgATP 2, NaGTP 2, phosphocreatine Na 2, EGTA 5, HEPES hemisodium 10 and lidocaine N-ethyl bromide 5 (QX-314) (pH 7.3; osmolarity 285–295 mOsmol/l). The LC cells were identified according to the IR-DIC image and then the cell attached configuration (3–8 GΩ seal resistance) was established. A brief negative pressure pulse was applied into the recording pipette to establish the whole-cell mode. Recordings of LC neurons were done 5 min after the rupture of the patch membrane for stabilizing the intracellular milieu. The acceptable access resistance of these recordings was < 20 MΩ during our experiments; otherwise, the data were excluded. Whole-cell voltage clamp recording was accomplished on LC slices using a MultiClamp 700B amplifier and pClamp 10 software (Molecular Devices, USA), with a 3-kHz low-pass Bessel filter and digitized at 10 kHz using a Digidata 1440A data acquisition system (Molecular Devices, Sunnyvale, CA, USA).
Recordings of evoked and spontaneous IPSCs were made in voltage-clamp mode at a holding potential of − 70 mV. IPSCs were pharmacologically isolated by bathing slices continuously with the AMPA receptor antagonist (CNQX; 20 µm) and NMDA receptor antagonist (AP-5; 50 µm). Bicuculline (20 µM), a GABAA receptor antagonist, was used to confirm GABAergic IPSCs. The amplitude and frequency of the spontaneous IPSCs were measured in LC slices from morphine-treated rats in the absence and presence of naloxone (1 µM). Recordings of the eIPSCs were performed by electrical stimulation through a concentric bipolar stimulating electrode located approximately 50–100 µm away from the recorded LC neurons. The stimulation intensity was gradually increased until we could detect an evoked IPSC [28]. The stimulation strength to record eIPSCs was set at 1.5 times above eIPSCs threshold stimuli (70–100 µA). In different experiments, IPSCs of LC slices were recorded during bath-application of orexin-A (100 nM), SB-334867 (3 µM; an OX1R antagonist) in the absence and presence of naloxone (1 µM; 10 min) [26, 29, 30]. Finally, in order to record paired-pulse ratio (PPR), paired pulses with 70-ms interval were delivered every 20 s. The PPR was calculated as the ratio of averaged amplitude of the second eIPSC (eIPSC2) to that of the first eIPSC (eIPSC1) [24].
Data analysis
Electrophysiological data are presented as mean ± SEM. Analyses of all data were done offline using Clampfit (pClamp 10; Molecular Devices) and MiniAnalysis (version 5.6.4; Synaptosoft, Decatur, GA) software. Statistical analysis was also done by GraphPad Prism version 7 for Windows (GraphPad Software, La Jolla, CA, USA). Behavioral symptoms were studied by Fisher’s exact probability test. Electrophysiological data were compared before and after drug application, using paired Student’s t test, one-sample t test, and repeated measures ANOVA or one-way ANOVA for multiple group comparisons. Newman–Keuls multiple comparison test was used following ANOVA to test significance among different groups. In this study, probability (P) values less than 0.05 were deemed statistically significant.
Results
Identification of locus coeruleus neurons
The LC neurons were identified under upright interference contrast microscopy as a dense and compact group of cells and also translucent area lying on the ventrolateral margin of the 4th ventricle [26, 31, 32] (Fig. 1a). In addition to morphological properties, some electrophysiological characteristics serve to distinguish these neurons from the adjacent neurons. These properties including the range of the resting membrane potentials from − 50 to − 60 mV, the rate of spontaneous discharges around 0.5–3/s and 70–80 mV in amplitude, rhythmic oscillations in membrane current at the frequency of 0.3–4/s and amplitudes ranging from 25 to 100 pA [33,34,35,36] (Fig. 1b).

Identification of LC neurons in LC slices. a Locus coeruleus nucleus was identified under infra-red microscopy as a dense and compact group of cells and translucent area lying on the ventrolateral margin of the 4th ventricle (bright areas). The LC neurons were prepared in a horizontal brainstem slice from a 14 to 21-day-old rat. b Representative samples showing intracellular recordings of identified LC neurons with the mean spontaneous activity of LC neurons of morphine-treated rats (0.2–4 Hz), resting membrane potential (− 51 ± 1.8 mV), and rhythmic oscillations. Representative traces of currents recorded from LC slices of the morphine-treated rats
The effect of naloxone on GABAergic IPSCs in LC neurons of control and morphine-treated rats
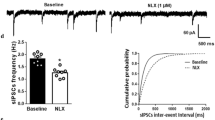
Evoked and spontaneous IPSCs were recorded in LC neurons from morphine-treated rats in the absence (baseline) and presence of naloxone. The amplitude of evoked IPSCs in LC neurons was decreased following naloxone application (1 µM) for 10 min (P < 0.05, Fig. 2a, b, n = 8). To investigate whether this effect was mediated by pre or postsynaptic mechanisms, we examined the effect of naloxone on the frequency and amplitude of spontaneous IPSCs in LC neurons. Naloxone (1 µM) significantly decreased sIPSC frequency (P < 0.05, Fig. 2d, n = 8) but did not change sIPSC amplitude compared to baseline (Fig. 2e). A rightward shift in the cumulative distribution of sIPSC inter-event intervals indicated a lower frequency of sIPSCs in LC neurons in the presence of naloxone (Fig. 2c, d). Other sIPSC parameters such as rise slope, decay time constant, and area under the curve were not statistically different in the absence and presence of naloxone. The average of the rise slope, decay time constant, and area under the curve of the sIPSCs in the absence of naloxone were (8.84 ± 1.86 pA/ms), (7.78 ± 1.12 ms), and (64.09 ± 12.50 pA/ms), respectively, and in the presence of naloxone were (4.08 ± 1.79 pA/ms), (5.19 ± 1.24 ms), and (56.06 ± 11.37 pA/ms), respectively.

Effect of naloxone on IPSCs in LC neurons of brain slices from morphine-treated rats. a Sample traces of eIPSCs at a holding potential of − 70 mV, in the absence (baseline) and presence of naloxone (1 µM, NLX). b Naloxone significantly decreased eIPSC amplitude (paired two-tailed Student’s t test, *P < 0.05, n = 8). c Sample traces of sIPSCs in the absence (baseline) and presence of naloxone. d Left, naloxone decreased sIPSCs frequency in LC neurons of morphine-treated rats (paired two-tailed Student’s t test, *P < 0.05, n = 8). Right, cumulative probability plots of sIPSCs inter-event interval in the absence (baseline) and presence of naloxone in LC neurons of morphine-treated rats. e Left, naloxone application did not change the sIPSCs amplitude of LC neurons of morphine-treated rats. Right, cumulative probability plots of sIPSCs amplitude in the absence (baseline) and presence of naloxone in LC neurons of morphine-treated rats
It is of interest to note that eIPSCs amplitude in the absence of naloxone (117.4 ± 6.147 pA) had no significant difference compared with naloxone administration (111.5 ± 7.272 pA) in control rats. In addition, sIPSCs frequency in the absence of naloxone (1.90 ± 0.057 Hz) did not show any significant difference compared with naloxone administration (1.78 ± 0.059 Hz) in control rats (n = 8; paired two-tailed Student’s t test; data not shown). These data indicate that the administration of naloxone had no significant difference on IPSCs when compared with baseline in control rats.
The effect of orexin-A on GABAergic IPSCs in LC neurons in the presence of naloxone
To assess whether OX1R activation enhanced the suppressive effect of naloxone on IPSCs, a series of experiments was performed on IPSCs in morphine-treated rats. As shown in Fig. 3, naloxone (1 µM) decreased significantly (P < 0.05) eIPSCs amplitude and sIPSCs frequency, this effect was more decreased significantly (P < 0.001) in the presence of orexin-A (100 nM) in LC neurons of morphine-treated rats. Moreover, to evaluate if SB-334867 prevented further reduction by co-application of naloxone plus orexin-A, we applied SB-334867 (3 µM) in the presence of naloxone plus orexin-A. SB-334867 reversed reduction effect of orexin-A on eIPSCs amplitude and sIPSCs frequency in the presence of naloxone. This finding indicated that orexin-A activation may induce further reduction IPSCs in morphine-treated rats in the presence of naloxone through OX1R.

OX1R activation mediated a further reduction in IPSCs through OX1R in the presence of naloxone. a Sample traces of eIPSCs at a holding potential of − 70 mV during bath application of naloxone, orexin-A and SB, respectively, without washout. b Evoked IPSCs amplitude decreased significantly (*P < 0.05) in the presence of naloxone (1 µM), this effect were more decreased significantly (***P < 0.001) in the presence of orexin-A (100 nM). SB-334867 (3 µM, SB) reversed the suppressive effect of orexin-A (100 nM) on eIPSCs amplitude (repeated measures ANOVA followed by Student–Newman–Keuls test, *P < 0.05, n = 8). c Sample traces of sIPSCs recorded during baseline and bath application of naloxone, orexin-A and SB, respectively, without washout. d Left, spontaneous IPSCs frequency decreased significantly (*P < 0.05) in the presence of naloxone (1 µM). This effect was more decreased significantly (***P < 0.001) in the presence of orexin-A (100 nM) (repeated measure ANOVA followed by Student–Newman–Keuls test, n = 8). This effect on sIPSCs frequency is reversed by SB-334867 (3 µM) treatment (repeated measure ANOVA followed by Student–Newman–Keuls test, *P < 0.05, n = 8). Right, cumulative probability plots of sIPSCs inter-event interval compared to baseline and bath application of naloxone, orexin-A and SB, respectively. e Left, sIPSCs amplitudes were not significantly different in baseline and bath application of naloxone, orexin-A and SB, respectively. Right, cumulative probability plots of sIPSCs amplitude during baseline and bath application of naloxone, orexin-A and SB, respectively
We next studied the effect of orexin-A on IPSCs in application order of orexin-A and naloxone, respectively, in LC neurons from morphine-treated rats. Application of orexin-A (100 nM) significantly decreased eIPSC amplitude of LC neurons in the presence of naloxone (1 µM) (P < 0.001, Fig. 4a, b, n = 8) but had no effect on baseline eIPSC amplitude when applied alone. Cumulative probability analysis of sIPSCs showed that orexin-A in the presence of naloxone also significantly reduced sIPSC frequency, as evidenced by the rightward shift of the inter-event intervals curve (Fig. 4c, d). Compared to baseline, orexin-A (100 nM) alone had no significant effect on sIPSC frequency (Fig. 4d) or amplitude (Fig. 4e). These results suggest that the GABAergic IPSCs were decreased with co-application of orexin-A plus naloxone in LC neurons from morphine-treated rats.

Orexin-A decreased IPSCs in LC neurons of morphine-treated rats in the presence of naloxone. a Sample traces of eIPSCs at a holding potential of − 70 mV in bath application of orexin-A (OXA) and orexin-A plus naloxone (NLX). b Orexin-A pretreatment (100 nM) had no effect on the eIPSC amplitude alone compared to baseline, while orexin-A reduced eIPSC amplitude in the presence of naloxone (1 µM) (repeated measures ANOVA followed by Student–Newman–Keuls test, *P < 0.001, ***P < 0.001, n = 8). c Sample traces of sIPSCs during baseline and bath application of orexin-A and orexin-A plus naloxone. d Left, orexin-A significantly reduced the frequency of sIPSCs in the presence but not in the absence of naloxone (repeated measure ANOVA followed by Student–Newman–Keuls test, *P < 0.05, ***P < 0.001, n = 8). Orexin-A alone had no significant effect on sIPSCs frequency compared to baseline. Right, cumulative probability plots of sIPSCs inter-event interval during baseline and bath application of orexin-A and orexin-A plus naloxone. e Left, orexin-A had no significant effect on sIPSCs amplitude either alone or with naloxone compared to baseline. Right, cumulative probability plots of sIPSCs amplitude during baseline and bath application of orexin-A and orexin-A plus naloxone
To identify the receptor mediating the effect of orexin-A on GABAergic IPSCs in the presence of naloxone, experiments were performed with the OX1R antagonist, SB-334867. While naloxone still led to a significant reduction in eIPSC amplitude (Fig. 5a, b, n = 8) and sIPSC frequency (Fig. 5c, d, n = 8), pretreatment with SB-334867 (3 µM) prevented further reduction by co-application of orexin-A plus naloxone (1 µM) (Fig. 5b, d). Again, no treatment tested had any effect on eIPSC amplitude (Fig. 5e).

Pretreatment with the OX1R antagonist, SB-334867, prevented the effect of orexin-A on GABAergic IPSCs in the presence of naloxone in LC neurons of morphine-treated rats. a Sample traces of eIPSCs at a holding potential of − 70 mV during bath application of SB, orexin-A and naloxone, respectively, without washout. b Evoked IPSCs in the presence of SB and SB plus orexin-A were not significantly different in the absence of naloxone (baseline). SB-334867 (3 µM, SB) reversed the suppressive effect of orexin-A (100 nM) on eIPSC amplitude in the presence of naloxone (1 µM) (repeated measures ANOVA followed by Student–Newman–Keuls test, *P < 0.05, n = 8), while SB-334867 had no significant effect on evoked IPSCs in the absence of naloxone (baseline). c Sample traces of sIPSCs recorded during baseline and bath application of SB, orexin-A and naloxone, respectively, without washout. d Left, SB-334867 reversed the orexin-A effect on sIPSCs frequency in the presence of naloxone, but not during baseline (repeated measure ANOVA followed by Student–Newman–Keuls test, *P < 0.05, n = 8). Right, cumulative probability plots of sIPSCs inter-event interval compared to baseline and bath application of SB and SB-orexin-A-naloxone, respectively, without washout. e Left, sIPSCs amplitudes were not significantly different in the absence (baseline) and the presence of naloxone. Right, cumulative probability plots of sIPSCs amplitude during baseline and bath application of SB and SB-orexin-A-naloxone, respectively
Furthermore, to determine whether LC neurons receive any endogenous orexinergic tone in our brain slices, we tested the effect of the OX1R antagonist, SB-334867 in the absence (baseline) and presence of naloxone. Application of SB-334867 (3 µM) alone did not significantly affect eIPSCs amplitude or sIPSCs frequency compared to baseline, nor did it reverse the suppressive effect of naloxone (3 µM) on either parameter (Fig. 6a–d, n = 8). In addition, SB-334867 had no effect on spontaneous IPSCs amplitude when applied alone or in combination with naloxone (Fig. 6e).

SB-334867 pretreatment had no significant effect on IPSCs in LC neurons of morphine-treated rats. a Sample traces of eIPSCs at a holding potential of − 70 mV in the presence of SB and SB plus naloxone. b SB-334867 pretreatment (3 µM) had no significant effect on eIPSCs in the absence of naloxone (repeated measure ANOVA followed by Student–Newman–Keuls test, *P < 05, n = 8) and no effect on the reduction in eIPSCs amplitude by naloxone. c Sample traces of sIPSCs during baseline and in the presence of SB and SB plus naloxone. d Left, SB-334867 pretreatment reduced sIPSCs frequency in the presence of naloxone compared to the absence of naloxone (baseline) (repeated measure ANOVA followed by Student–Newman–Keuls test, *P < 0.05, n = 8). Right, cumulative probability plots of the sIPSC inter-event interval during baseline and bath application of SB and SB plus naloxone. e Left, SB-334867 pretreatment had no significant effect on sIPSCs amplitude in either the absence or presence of naloxone compared to baseline. Right, cumulative probability plots of sIPSCs amplitude during baseline and bath application of SB and SB plus naloxone
Finally, in order to investigate whether the co-application of orexin-A and naloxone reduced GABAergic IPSCs in LC neurons of morphine-treated rats more, we compared the effect of orexin-A plus naloxone on IPSC versus naloxone alone (Fig. 7a). These data were expressed as a percentage of decrease in eIPSCs amplitude and sIPSCs frequency from baseline.

Co-application of orexin-A plus naloxone more reduced GABAergic IPSCs in LC neurons of morphine-treated rats. a Statistical comparison of eIPSCs amplitude between naloxone alone, co-application of orexin-A plus naloxone, and co-application of SB-orexin-A-naloxone vs. orexin-A alone. b Statistical comparison of sIPSCs frequency between naloxone alone, co-application of orexin-A plus naloxone, and co-application of SB-orexin-A-naloxone vs. orexin-A alone. The application of orexin-A (100 nM) alone in the absence of naloxone had no significant effect on eIPSCs amplitude and sIPSCs frequency (one-sample t test). Naloxone alone significantly (P < 0.05) decreased eIPSCs amplitude (11.89 ± 4.57%) and sIPSCs frequency (16.76 ± 5.05%) compared with orexin-A. Co-application of orexin-A plus naloxone on eIPSCs amplitude (32.90 ± 5.26%) and sIPSCs frequency (42.40 ± 4.53%) vs. orexin-A alone had significant difference (P < 0.001). Co-application of SB-orexin-A-naloxone also significantly (P < 0.05) decreased eIPSCs amplitude (8.12 ± 1.39%) and sIPSCs frequency (12.13 ± 2.04%) compared with orexin-A alone. In addition, percentage of decrease in eIPSCs amplitude (21.01 ± 5.13%) and sIPSCs frequency (25.64 ± 5.58%) was significant (P < 0.05) between co-application orexin-A and naloxone vs. naloxone alone. Each bar represents the mean ± SEM (*) indicated statistical significance between orexin-A alone compared to naloxone alone and co-application of orexin-A and naloxone (P < 0.05, P < 0.001) and also (#) signified statistical significance for comparison between naloxone alone and co-application of orexin-A and naloxone (P < 0.05)
However, since the application of orexin-A (100 nM) alone in the absence of naloxone had no significant effect on IPSCs compared to baseline, these comparisons were performed with orexin-A alone (Fig. 7a, b). A one-sample t test was applied to confirm that the slight decrease of eIPSCs amplitude and sIPSCs frequency were not significant changes. The reduction in eIPSC amplitude (21.01 ± 5.13%) and sIPSC frequency (25.64 ± 5.58%) by co-application of orexin-A and naloxone was significantly greater than the application of naloxone alone (P < 0.05, Fig. 7a, b, n = 8). Furthermore, the effect of co-application of SB-334867 and orexin-A on GABAergic IPSCs in the presence of naloxone showed that pretreatment with SB-334867 (3 µM) did prevent further reduction by orexin-A in the presence of naloxone (1 µM) and had no significant difference compared with naloxone alone. These results suggest that orexin-A may enhance the suppressive effect of naloxone on GABAergic IPSCs through a reduction in presynaptic GABA release probability.
To further confirm whether the orexin-A effect on IPSCs with another MOR antagonist, were performed an additional experiment using the selective μ-receptor antagonist, CTAP (1 μM). Orexin-A had no significant effect on eIPSCs amplitude in the absence of CTAP, while eIPSC amplitude is decreased in the presence of CTAP in LC neurons of morphine-treated rats [(P < 0.001); Fig. 8a, b, n = 8]. Similarly, orexin-A had no significant effect on sIPSCs frequency in the absence of CTAP, but sIPSCs frequency is significantly decreased in the presence of CTAP. Moreover, orexin-A had no significant effect on sIPSCs amplitude in the absence and presence of CTAP (Fig. 8c–e, n = 8). Pretreatment with SB-334867 (3 μM), prevented the effect of orexin-A eIPSCs amplitude on and sIPSCs frequency in the presence of CTAP in LC neurons of morphine-treated rats (data not shown).

Orexin-A decreased IPSCs in LC neurons of morphine-treated rats in the presence of CTAP. a Sample traces of eIPSCs in bath application of orexin-A (OXA) and orexin-A plus CTAP. b Orexin-A pretreatment (100 nM) had no effect on the eIPSC amplitude alone compared to baseline, while orexin-A reduced eIPSC amplitude in the presence of CTAP (1 µM) (repeated measures ANOVA followed by Student–Newman–Keuls test, ***P < 0.001, n = 8). c Sample traces of sIPSCs during baseline and bath application of orexin-A and orexin-A plus CTAP. d Left, orexin-A significantly reduced the frequency of sIPSCs in the presence but not in the absence of CTAP (repeated measure ANOVA followed by Student–Newman–Keuls test, ***P < 0.001, n = 8). Orexin-A alone had no significant effect on sIPSCs frequency compared to baseline. Right, cumulative probability plots of sIPSCs inter-event interval during baseline and bath application of orexin-A and orexin-A plus CTAP. e Left, orexin-A had no significant effect on sIPSCs amplitude either alone or with CTAP compared to baseline. Right, cumulative probability plots of sIPSCs amplitude during baseline and bath application of orexin-A and orexin-A plus CTAP
Orexin-A suppressed eIPSCs in the presence of naloxone in LC neurons of morphine-treated rats via a presynaptic mechanism
To more elucidate whether pre or post-synaptic mechanism involved in the orexin-A-induced eIPSC suppression, we evaluated the effect of orexin-A on the paired pulse ratio (PPR) of paired eIPSCs. It was established that PPR alterations are related to presynaptic modulation [37]. Orexin-A increased significantly the PPR of paired eIPSCs amplitude in the presence of naloxone (1 µM) (one-way ANOVA, P < 0.05, Fig. 9a, b) and this effect is reversed by SB (3 μM) (data not shown). These results suggest that orexin-A inhibits GABAergic IPSCs in LC neurons of morphine-treated rats through the presynaptic mechanism by inhibiting evoked GABA release.

Orexin-A increased the PPR of paired IPSCs in the presence of naloxone in LC neurons of morphine-treated rats. a Paired IPSCs were evoked by 70 ms-separated paired pulses every 20 s. b Averaged PPR, the ratio of the averaged amplitude of 2nd IPSCs to that of 1st IPSCs in the presence of orexin-A (100 nM) and naloxone (1 μM) (data are shown as mean ± SEM, one-way ANOVA, **P < 0.01 versus baseline, n = 8)
Discussion
In this report, we used the application of naloxone to brainstem slices of morphine-treated rats as an in vitro model to study the synaptic effects of acute opioid withdrawal. Using this model, we show that naloxone reduced GABAergic IPSCs in LC neurons. Furthermore, we found that orexin-A, via its receptor OX1R, enhanced the suppressive effect of naloxone on IPSCs in LC neurons suggests that orexinergic signaling to the LC also modulates GABAergic neurotransmission but only in the context of withdrawal. The reduction in the efficiency of GABAergic inhibitory synaptic transmission in LC neurons of morphine-treated rats by orexin-A in the presence of naloxone suggests this could be a mechanism for driving LC activity during acute morphine withdrawal.
Evoked GABAergic IPSC amplitudes were significantly reduced following naloxone application to LC neurons of morphine-treated rats; an effect that may be due to a reduction in the postsynaptic response to GABA and/or a decrease in presynaptic GABA release. We further observed that naloxone also decreased spontaneous IPSC frequency in LC neurons, but had no effect on spontaneous IPSCs amplitude. Our finding of a rightward shift of the inter-event interval histogram of spontaneous IPSCs supports the conclusion that naloxone suppressed GABAA-mediated IPSCs in LC neurons via a presynaptic mechanism and that LC neuron firing activity may therefore be enhanced by the decrease in GABAergic input during naloxone-induced morphine withdrawal.
Our data are comparable with previous studies showing that endogenously released adenosine inhibits GABA release during morphine withdrawal through presynaptic mechanism [29]. Behaviorally, microinjection of the GABAA receptor agonist, muscimol, into the LC attenuated the signs of naloxone-induced withdrawal in morphine-treated rats [25]. In contrast, it has been reported that the probability of GABA release was increased during withdrawal in the dopamine neurons of guinea pig midbrain [38], and that GABAergic currents are increased during naloxone morphine withdrawal in ventral tegmental area (VTA) [39]. It is therefore likely that chronic morphine exposure drives multiple and complex changes to GABAergic neurotransmission throughout the brain depending on exposure time, dosing and the region examined [40].
Considering that noradrenergic nuclei are involved in the state of opioid withdrawal [41], another electrophysiological study also suggested that noradrenaline signaling initiates GABAA inhibition in the stria terminalis during acute opioid withdrawal [42]. It was previously reported that LC activity was increased during morphine withdrawal syndrome [3]. We know that cAMP signaling plays an important role in morphine withdrawal in this nucleus [43] and up-regulation of adenylyl cyclase-cAMP signaling regulates presynaptic GABA release [3]. It has been shown that adenosine receptors exert an effect on GABA release via other neuromodulators like D1 dopamine receptors in striatonigral neurons [43,44,45,46]. Taken together, it could be concluded that naloxone application can reduce IPSCs and subsequently increase LC activity in the morphine-treated rats.
We also found that orexin-A likely enhanced the suppressive effect of naloxone GABAA-mediated IPSCs in LC neurons of morphine-treated rats. Based on results, it is suggested that orexin-A reduces IPSCs by decreasing presynaptic GABA release probability. Although orexinergic fibers can have a direct effect on LC neurons [10], orexin-A alone had no significant effect on IPSCs compared to baseline. This suggests either that the mechanism of orexin-mediated inhibition of GABAergic transmission is indirect, or that it normally masked by MOR activity and requires the presence of naloxone to be revealed. Therefore, these effects are due to morphine dependency in rats and same effects are probably to be different in control rats.
It has been proposed that the protein kinase C (PKC) has a role in modification of the expression of naloxone-precipitated withdrawal signs [47]. Several lines of evidence indicate that PKC activated by increased calcium plays an important role in the orexinergic system activity [48, 49]. An electrophysiological study showed that the naloxone injection increased the cAMP concentration in LC neurons of morphine-treated rats and this effect was reduced by OX1R blockade [17]. Moreover, orexin-A via OX1R also potentiated noradrenaline release through PKC signaling pathway in the LC neurons [50]. Moreover, PKC has a critical effect in orexin-A signaling, as observed previously in the VTA [49]. In addition, PKC is an important modulator of the processes of morphine tolerance and dependence operating via numerous signaling cascades through the phosphorylation of target proteins [51]. Exposure to orexin-A decreased the amplitude of the GABAergic currents through PKC and CaMKII phosphorylation in transfected HEK293 cell [52]. Taken together, this suggests that this effect of orexin-A on eIPSCs could be mediated by PKC in the presence of naloxone. Further work will be necessary to investigate these and other potential orexin-A signaling mechanisms resulting in a reduction in inhibitory neurotransmission in the context of naloxone-induced opioid withdrawal in LC neurons.
Our previous behavioral findings have shown that symptoms of naloxone-induced opioid withdrawal in vivo can be attenuated by pre-treatment with the OX1R antagonist SB-334867 given directly to the LC [20]. In addition, it is reported that morphine withdrawal increases mRNA expression of orexin-A in the lateral hypothalamus projections [42], which are found at high density in the LC [9]. Therefore, we hypothesized that OX1R antagonism would reverse the naloxone-induced reduction in GABAergic neurotransmission. To our surprise, however, measurement of eIPSC amplitude in LC neurons during pretreatment with the OX1R antagonist, SB-334867, in the presence of naloxone showed no difference compared to naloxone application alone, suggesting that LC neurons did not receive any endogenous orexinergic tone in our brain slices. It is possible that these projections have simply been severed during the brain slice preparation and are no longer able to release orexin. Detailed tracing of orexinergic projections to the LC may help to elucidate their specific role in the modulation of GABAergic transmission during morphine withdrawal.
Conclusions
Our data provide direct electrophysiological evidence that naloxone reduces GABAergic neurotransmission in LC neurons of morphine-treated rats. The major finding of this study is that orexin-A via OX1R further decreased GABA release in the presence of naloxone via a presynaptic mechanism. We conclude that orexin-A, via indirect modulation of GABAA-mediated IPSCs, may have a critical role in regulating the excitability of LC neurons in the context of morphine withdrawal. Further understanding of the orexinergic system may therefore be fruitful for the development of therapies for the treatment of morphine dependency.
References
Koob GF, Maldonado R, Stinus L (1992) Neural substrates of opiate withdrawal. Trends Neurosci 15:186–191
Aghajanian GK (1978) Tolerance of locus coeruleus neurones to morphine and suppression of withdrawal response by clonidine. Nature 276(5684):186–188
Rasmussen K, Beitner-Johnson DB, Krystal JH, Aghajanian GK, Nestler EJ (1990) Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci 10(7):2308–2317
Maldonado R, Stinus L, Gold LH, Koob GF (1992) Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Ther 261(2):669–677
Aston-Jones G, Chiang C, Alexinsky T (1991) Discharge of noradrenergic locus coeruleus neurons in behaving rats and monkeys suggests a role in vigilance. Prog Brain Res 88:501–520
Nestler EJ, Hope BT, Widnell KL (1993) Drug addiction: a model for the molecular basis of neural plasticity. Neuron 11:995–1006
Stamford JA (1995) Descending control of pain. Br J Anaesth 75:217–227
Sara SJ (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10:211–223
Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K (1999) Distribution of orexin neurons in the adult rat brain. Brain Res 827:243–260
Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, Benham CD, Taylor SG, Routledge C, Hemmati P et al (1999) Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci USA 96:10911–10916
Peyron C, Tighe DK, Van den Pol A, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS (1998) Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18:9996–10015
Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S et al (1998) Orexins and orexin receptors, a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–585
Hervieu GJ, Cluderay JE, Harrison DC, Roberts JC, Leslie RA (2001) Gene expression and protein distribution of the orexin-1 receptor in the rat brain and spinal cord. Neuroscience 103:777–797
Azizi H, Mirnajafi-Zadeh J, Rohampour K, Semnanian S (2010) Antagonism of orexin type 1 receptors in the locus coeruleus attenuates signs of naloxone-precipitated morphine withdrawal in rats. Neurosci Lett 482:255–259
Mousavi Y, Azizi H, Mirnajafi-Zadeh J, Javan M, Semnanian S (2014) Blockade of orexin type-1 receptors in locus coeruleus nucleus attenuates the development of morphine dependency in rats. Neurosci Lett 578:90–94
Ahmadi-Soleimani SM, Azizi H, Gompf HS, Semnanian S (2017) Role of orexin type-1 receptors in paragiganto-coerulear modulation of opioid withdrawal and tolerance: a site specific focus. Neuropharmacology 126:25–37
Fakhari M, Azizi H, Semnanian S (2017) Central antagonism of orexin type-1 receptors attenuates the development of morphine dependence in rat locus coeruleus neurons. Neuroscience 363:1–10
Georgescu D, Zachariou V, Barrot M, Mieda M, Willie JT, Eisch AJ, Yanagisawa M, Nestler EJ, DiLeone RJ (2003) Involvement of the lateral hypothalamic peptide orexin in morphine dependence and withdrawal. J Neurosci 23:3106–3111
Sharf R, Sarhan M, Dileone RJ (2008) Orexin mediates the expression of precipitated morphine withdrawal and concurrent activation of the nucleus accumbens shell. Biol Psychiatry 64:175–183
Davoudi M, Azizi H, Mirnajafi-Zadeh J, Semnanian S (2016) The blockade of GABAA receptors attenuates the inhibitory effect of orexin type 1 receptors antagonist on morphine withdrawal syndrome in rats. Neurosci Lett 617:201–206
Hooshmand B, Azizi H, Javan M, Semnanian S (2017) Intra-LC microinjection of orexin type-1 receptor antagonist SB-334867 attenuates the expression of glutamate-induced opiate withdrawal like signs during the active phase in rats. Neurosci Lett 636:276–281
Ghaemi-Jandabi M, Azizi H, Ahmadi-Soleimani SM, Semnanian S (2017) Intracoerulear microinjection of orexin-A induces morphine withdrawal-like signs in rats. Brain Res Bull 130:107–111
Dergacheva O, Philbin R, Bateman P, Mendelowitz D (2012) Orexinergic modulation of GABAergic neurotransmission to cardiac vagal neurons in the brain stem nucleus ambiguus changes during development. Neuroscience 209:12–20
Ho YC, Lee HJ, Tung LW, Liao YY, Fu SY, Teng SF, Liao HT, Mackie K, Chiou LC (2011) Activation of orexin 1 receptors in the periaqueductal gray of male rats leads to antinociception via retrograde endocannabinoid (2-arachidonoylglycerol)-induced disinhibition. J Neurosci 31:14600–14610
Mirzaii-Dizgah I, Karimian SM, Hajimashhadi Z, Riahi E, Ghasemi T (2008) Attenuation of morphine withdrawal signs by muscimol in the locus coeruleus of rats. Behav Pharmacol 19(3):171–175
Kaeidi A, Azizi H, Javan M, Ahmadi-Soleimani SM, Fathollahi Y, Semnanian S (2015) Direct facilitatory role of paragigantocellularis neurons in opiate withdrawal-induced hyperactivity of rat locus coeruleus neurons: an in vitro study. PLoS One 10(7):e0134873
Jones KL, Barr GA (1995) Ontogeny of morphine withdrawal in the rat. Behav Neurosci 109:1189–1198
Pan YZ, Li DP, Chen SR, Pan HL (2002) Activation of delta-opioid receptors excites spinally projecting locus coeruleus neurons through inhibition of GABAergic inputs. J Neurophysiol 88(5):2675–2683
Hack SP, Vaughan CW, Christie MJ (2003) Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology 45:575–584
Rohampour K, Azizi H, Fathollahi Y, Semnanian S (2017) Peripheral nerve injury potentiates excitatory synaptic transmission in locus coeruleus neurons. Brain Res Bull 130:112–117
Belujon P, Baufreton J, Grandoso L, Boué-Grabot E, Batten TF, Ugedo L, Garret M, Taupignon AI (2009) Inhibitory transmission in locus coeruleus neurons expressing GABAA receptor epsilon subunit has a number of unique properties. J Neurophysiol 102(4):2312–2325
Mohammad Ahmadi Soleimani S, Azizi H, Pachenari N, Mirnajafi-Zadeh J, Semnanian S (2017) Enhancement of μ-opioid receptor desensitization by orexin-A in rat locus coeruleus neurons. Neuropeptides 63:28–36
Williams JT, Marshall KC (1987) Membrane properties and adrenergic responses in locus coeruleus neurons of young rats. J Neurosci 7:3687–3694
Christie MJ, Williams JT, North RA (1989) Electrical coupling synchronizes subthreshold activity in locus coeruleus neurons in vitro from neonatal rats. J Neurosci 9:3584–3589
Shen KZ, North RA (1992) Muscarine increases cation conductance and decreases potassium conductance in rat locus coeruleus neurones. J Physiol 455:471–485
Van den Pol AN, Ghosh PK, Liu R-J, Li Y, Aghajanian GK, Gao X-B (2002) Hypocretin (orexin) enhances neuron activity and cell synchrony in developing mouse GFP-expressing locus coeruleus. J Physiol 541:169–185
Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405
Chieng B, Williams JT (1998) Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci 18(17):7033–7039
Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ (1998) Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci 18:10269–10276
Yanez A, Sabbe MB, Stevens CW, Yaksh TL (1990) Interaction of midazolam and morphine in the spinal cord of the rat. Neuropharmacology 29(4):359–364
Maldonado R (1997) Participation of noradrenergic pathways in the expression of opiate withdrawal: biochemical and pharmacological evidence. Neurosci Biobehav Rev 21(1):91–104
Zhou Y, Bendor J, Hofmann L, Randesi M, Ho A, Kreek MJ (2006) Mu opioid receptor and orexin/hypocretin mRNA levels in the lateral hypothalamus and striatum are enhanced by morphine withdrawal. J Endocrinol 191:137–145
Madhavan A, He L, Stuber GD, Bonci A, Whistler JL (2010) Micro-opioid receptor endocytosis prevents adaptations in ventral tegmental area GABA transmission induced during naloxone-precipitated morphine withdrawal. J Neurosci 30:3276–3286
Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ (1991) A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res 548:100–110
Floran B, Barajas C, Floran L, Erlij D, Aceves J (2002) Adenosine A1 receptors baseline dopamine D1-dependent [3H] GABA release in slices of substantia nigra pars reticulata and motor behavior in the rat. Neuroscience 115(3):743–751
Chakrabarti S, Oppermann M, Gintzler AR (2001) Chronic morphine induces the concomitant phosphorylation and altered association of multiple signaling proteins: a novel mechanism for modulating cell signaling. Proc Natl Acad Sci USA 98(7):4209–4214
Ohsawa M, Kamei J (1999) Modification of the expression of naloxone-precipitated withdrawal signs in morphine-dependent mice by diabetes: possible involvement of protein kinase C. Jpn J Pharmacol 79(3):303–311
Lund PE, Shariatmadari R, Uustere A, Detheux M, Parmentier M, Kukkonen JP, Akerman KE (2000) The orexin OX1 receptor activates a novel Ca2+ influx pathway necessary for coupling to phospholipase C. J Biol Chem 275(40):0806–30812
Bailey CP, Kelly E, Henderson G (2004) Protein kinase C activation enhances morphine-induced rapid desensitization of µ-opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol 66:1592–1598
Chen XW, Mu Y, Huang HP, Guo N, Zhang B, Fan SY, Xiong JX, Wang SR, Xiong W, Huang W, Liu T, Zheng LH, Zhang CX, Li LH, Yu ZP, Hu ZA, Zhou Z (2008) Hypocretin-1 potentiates NMDA receptor-mediated somatodendritic secretion from locus ceruleus neurons. J Neurosci 28(12):3202–3208
Bonci A, Williams J (1997) Increased probability of GABA release during withdrawal from morphine. J Neurosci 17(2):796–803
Sachidanandan D, Reddy HP, Mani A, Hyde GJ, Bera AK (2017) The neuropeptide orexin-A inhibits the GABAA receptor by PKC and Ca2+/CaMKII-dependent phosphorylation of its β1 subunit. J Mol Neurosci 61(4):459–467
Acknowledgements
This work was supported by the Cognitive Sciences and Technologies Council of Iran (CSTC, Grant no. 95P31), Iran National Science Foundation (INSF) and the Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran. The authors also wish to thank Dr. Alexander Davies for his English correction of this manuscript and Dr. Amir Shojaei for his technical help.
Author information
Authors and Affiliations
Corresponding authors
About this article

Cite this article
Davoudi, M., Azizi, H., Mirnajafi-Zadeh, J. et al. Decrease of inhibitory synaptic currents of locus coeruleus neurons via orexin type 1 receptors in the context of naloxone-induced morphine withdrawal. J Physiol Sci 69, 281–293 (2019). https://doi.org/10.1007/s12576-018-0645-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-018-0645-1