
Abstract
Background
Galactosemia due to complete or near-complete galactose-1-phosphate uridyltransferase (GALT) deficiency was the first disorder added to the pioneering newborn screening panel besides phenylketonuria. In the last 50 years, many criticisms have been focused on the opportunity of its inclusion. Consequently, long-term single center experiences with this issue are generally lacking.
Methods
We reviewed the outcome of newborn screening for hypergalactosemia performed at our department since 1982 and the correspondent long-term clinical outcome.
Results
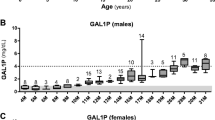
Among 1 123 909 newborns screened for hypergalactosemia, 33 showed abnormal results confirmed at second tier test. Thirteen patients were affected with classic galactosemia, 8 partial GALT deficiency, 3 severe galactokinase deficiency, 7 transient galactosemia, one congenital porto-systemic shunt, and one glucose transporter 2 deficiency. Acute neonatal liver failure in the late first week of life (5.8±1.1 days) unavoidably complicated the clinical course of classic galactosemia, unless in three second-born siblings treated on the basis of presumptive diagnosis immediately after newborn screening sample collection on day 3. Despite early treatment and long-term steadily normal peripheral blood galactose, 77% of patients with severe GALT deficiency present mild to severe intellectual disabilities. All patients with partial GALT deficiency showed normal intellectual development on a regular diet, as well as patients with galactokinase deficiency under treatment.
Conclusions
Availability of screening results within the fifth day after birth would allow the prevention of acute decompensation in classic galactosemia. A systematic diagnostic work-up in all positive newborns is essential to unravel the etiology of hypergalactosemia.
Similar content being viewed by others


References
Guthrie R. Screening for “inborn errors of metabolism” in the newborn infant-a multiple test program. Birth Defects Orig Artic Ser 1968;IV:92–98.
Schweitzer S. Newborn mass screening for galactosemia. Eur J Pediatr 1995;154:S37–S39.
Ridel KR, Leslie ND, Gilbert DL. An updated review of the long-term neurological effects of galactosemia. Pediatr Neurol 2005;33:153–161.
Fernandes J, Saudubray JM, van den Berghe G, Walter JH. Inborn metabolic diseases: diagnosis and treatment, 4th ed. Heidelberg: Springer, 2006.
Misuma H, Wada H, Kawakami M, Ninomiya H, Shohmori T. Galactose and galactose-1-phosphate spot test for galactosemia screening. Clin Chim Acta 1981;111:27–32.
Vaccaro Torracca AM. Galactosemia: qualitative and quantitative identification of galactose-1-phosphate. Istisan 1979;91:19.
Peduto A, Spada M, Alluto A, La Dolcetta M, Ponzone A, Santer R. A novel mutation in the GLUT2 gene in a patient with Fanconi-Bickel syndrome detected by neonatal screening for galactosaemia. J Inherit Metab Dis 2004;27:279–280.
Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E. Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis 2011;34:357–366.
Potter NL, Nievergelt Y, Shriberg LD. Motor and speech disorders in classic galactosemia. JIMD Rep 2013;11:31–41.
Plass AM, van El CG, Pieters T, Cornel MC. Neonatal screening for treatable and untreatable disorders: prospective parents’ opinions. Pediatrics 2010;125:e99–e106.
Berry GT. Galactosemia: when is it a newborn screening emergency? Mol Genet Metab 2012;106:7–11.
Coman DJ, Murray DW, Byrne JC, Rudd PM, Bagaglia PM, Doran PD, et al. Galactosemia, a single gene disorder with epigenetic consequences. Pediatr Res 2010;67:286–292.
Ficicioglu C, Thomas N, Yager C, Gallagher PR, Hussa C, Mattie A, et al. Duarte (DG) galactosemia: a pilot study of biochemical and neurodevelopmental assessment in children detected by newborn screening. Mol Genet Metab 2008;95:206–212.
Powell KK, Van Naarden Braun K, Singh RH, Shapira SK, Olney RS, Yeargin-Allsopp M. Long-term speech and language developmental issues among children with Duarte galactosemia. Genet Med 2009;11:874–879.
Janzen N, Illsinger S, Meyer U, Shin YS, Sander J, Lücke T, et al. Early cataract formation due to galactokinase deficiency: impact of newborn screening. Arch Med Res 2011;42:608–612.
Ferrero GB, Porta F, Biamino E, Mussa A, Garelli E, Chiappe F, et al. Remittent hyperammonemia in congenital portosystemic shunt. Eur J Pediatr 2010;169:369–372.
Kim MJ, Ko JS, Seo JK, Yang HR, Chang JY, Kim GB, et al. Clinical features of congenital portosystemic shunt in children. Eur J Pediatr 2012;171:395–400.
Santer R, Steinmann B, Schaub J. Fanconi-Bickel syndrome—a congenital defect of facilitative glucose transport. Curr Mol Med 2002;2:213–227.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Porta, F., Pagliardini, S., Pagliardini, V. et al. Newborn screening for galactosemia: a 30-year single center experience. World J Pediatr 11, 160–164 (2015). https://doi.org/10.1007/s12519-015-0017-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-015-0017-3