
Abstract
Introduction
We aimed to describe healthcare resource utilization (HCRU) patterns and costs in patients with fibrosing interstitial lung disease (ILD) and those with a progressive phenotype of fibrosing ILD in a US claims database.
Methods
Data from the IBM® MarketScan® databases (1 October 2011–30 September 2015) were used. Diagnosis codes documented on medical claims on two occasions (without any claims during the 12 months prior) identified patients with incident fibrosing ILD. Patients with chronic fibrosing ILD with a progressive phenotype were identified by proxies for progression. Patients aged ≥ 18 years with 365 days of continuous coverage before the index date were eligible for inclusion. Data were analyzed for 12 months prior to identification of fibrosing ILD/progressive phenotype (baseline) and 12 months after (follow-up). Outcomes included treatment patterns, outpatient and inpatient claims, and costs.
Results
We identified 23,577 patients with incident fibrosing ILD and 14,722 with the progressive phenotype. Follow-up data were available for 9986 and 5840 patients, respectively. The most frequent ILD-related medications during baseline were corticosteroids (49.4% and 56.6%). Mean (± standard deviation [SD]) annualized number of outpatient claims was 30.0 (± 26.4) and 34.1 (± 27.7) in the baseline period and 36.2 (± 28.6) and 41.9 (± 30.2) in the follow-up in fibrosing ILD and with a progressive phenotype, respectively. Mean (SD) number of all-cause hospitalizations was 0.5 (± 1.1) and 0.7 (± 1.2) during baseline and 0.6 (± 1.1) and 0.7 (± 1.2) during follow-up. Mean (SD) total costs were $40,907 (± 92,496) and $49,561 (± 98,647) during baseline and $46,157 (± 102,858) and $54,215 (± 116,833) during follow-up. Inpatient mortality during follow-up was 53.50 and 77.44 per 1000 patient-years.
Conclusion
HCRU and costs were high in patients with chronic fibrosing ILD with a progressive phenotype, likely reflecting the disease severity and the need for close monitoring and acute care. Outpatient claims accounted for a substantial proportion of the total costs.
Plain Language Summary
Some patients with lung diseases have inflammation or scarring of the lung tissues (interstitial lung diseases, or ILDs). In some patients with lung scarring, the scarring may become progressive (i.e., it worsens over time). In this study, we looked at these patients identified in US health insurance records. We counted how many times patients visited a doctor, were admitted to hospital, or needed medications or tests. We also looked at the total cost of all this medical care. Overall, we concluded that patients with ILDs with progressive lung scarring had a high number of visits to the doctor, and the total costs of their medical care were high.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Patients with chronic fibrosing interstitial lung disease (ILD) other than idiopathic pulmonary fibrosis (IPF) may develop a progressive phenotype. |
There are currently limited data available on healthcare resource utilization (HCRU) and costs associated with non-IPF fibrosing ILD with a progressive phenotype. |
The present study aimed to describe HCRU patterns and costs in patients with fibrosing ILD and those with a progressive phenotype of fibrosing ILD in a US claims database. |
What was learned from the study? |
HCRU and costs were high in patients with chronic fibrosing ILD with a progressive phenotype, likely reflecting the disease severity and the need for close monitoring and acute care. |
Progressive disease may be associated with greater total healthcare resource use and costs than fibrosing ILD that is not progressive. |
Introduction
Interstitial lung diseases (ILDs) comprise a group of diffuse parenchymal lung diseases that may be of unknown cause (such as in the case of idiopathic pulmonary fibrosis [IPF]) or associated with a range of different disorders [1]. Some patients with chronic fibrosing ILD develop a progressive phenotype, characterized by worsening lung function and symptoms and increasing fibrosis of the lungs [2, 3]. Patients with ILD other than IPF, such as those associated with connective tissue diseases, sarcoidosis or chronic hypersensitivity pneumonitis, may also develop a progressive fibrosing phenotype with similar clinical characteristics to IPF [2].
Nintedanib and pirfenidone were approved for the treatment of IPF in the US in 2014 [4, 5], but until recently there were no approved treatments to slow other progressive fibrosing ILDs. Nintedanib is approved to slow lung function decline in systemic sclerosis-associated ILD and is also now approved for the treatment of patients with chronic fibrosing ILDs with a progressive phenotype [4]. Corticosteroids and immunosuppressants are commonly prescribed to patients with these conditions, but there is a lack of evidence to support their efficacy [6], and they are associated with side effects, including infection. As the treatment landscape is now changing dramatically, there is a need to better understand the characteristics of patients with chronic fibrosing ILDs with a progressive phenotype, what comorbidities they may have, how they are currently managed and treated, and their healthcare resource utilization (HCRU) and costs. This information is key for payers and healthcare decision-makers to optimize allocation of resources, ensure timely diagnostic assessment, and support effective management of this patient population.
Consideration of fibrosing ILDs with different etiologies but a progressive phenotype as a related group is a relatively new concept [7, 8]. Chronic respiratory diseases are among the most expensive health conditions in the US, accounting for > $100 billion in annual spending [9]. Although it has been postulated that the burden and healthcare costs for patients with fibrosing ILD with a progressive phenotype other than IPF are similar to those for patients with IPF [10], there are currently limited data available about HCRU. A US-based study, using both a different insurance claims database and algorithm, found that patients with ILD with frequent pulmonary visits (used as a proxy for progressive disease) had higher annual medical costs and more hospital claims than patients with ILD without frequent pulmonary visits [11]. However, data on diagnostic tests, medications, or clinical outcomes were not included. Healthcare utilization in patients with fibrosing ILDs with a progressive phenotype is likely to be high and affected by the following factors [10]: challenging diagnosis requiring a large number of tests and potentially repeated testing; close monitoring of disease, including regular clinic visits; therapies including pharmacologic treatment and supplemental oxygen; care of comorbidities such as cardiovascular disease and gastroesophageal reflux; and hospitalizations and emergency room visits.
We conducted a study estimating the prevalence and incidence of chronic fibrosing ILD with a progressive phenotype in the IBM® MarketScan® Commercial Claims and Encounters and Medicare Supplemental databases using an algorithm developed in collaboration with clinical experts [12]; these patients were used in the present study to investigate HCRU. The aim of the present study was to describe patients before and for 365 days after identification of fibrosing ILD or a fibrosing ILD with a progressive phenotype in terms of HCRU (outpatient and inpatient claims and medications) and costs (total, outpatient, inpatient, pharmacy, diagnostic tests). We also describe clinical characteristics, comorbidities in the baseline period, medical conditions during the follow-up period, and inpatient mortality during follow-up.
Methods
Study Design and Data Source
This study used data from the IBM® MarketScan® Commercial Claims and Encounters and Medicare Supplemental databases (1 October 2011–30 September 2015). As previously described [12], diagnosis codes from medical claims (International Classification of Diseases, Ninth Revision, Clinical Modification [ICD-9 CM]) [13] were used to identify patients with fibrosing ILD. Proxy measures based on codes for procedures (Current Procedural Terminology, 4th Edition), prescriptions (National Drug Codes and Healthcare Common Procedure Coding System), and other resource utilization were used to identify those with a progressive phenotype (detailed below). The use of the de-identified data was approved for exemption by the New England Independent Review Board.
Patients
Incident fibrosing ILD diagnosis was defined as a new lung fibrosis claim without a claim during the 365 days prior and followed by a second claim within 30–365 days, using the second claim as the index date (Fig. 1). Eligible ICD-9 CM codes for a diagnosis of fibrosing ILD are shown in Supplementary Table S1. Patients were aged ≥ 18 years at index date and were required to have 365 days of continuous medical and pharmacy insurance coverage prior to the index date (baseline period). Gaps of up to 30 days in coverage were permitted. If a patient had multiple separate periods of enrollment with fibrosing ILD claims, they were only included the first time they qualified.

Definition of a incident fibrosing ILD and b incident progressive fibrosing ILD. HCRU healthcare resource utilization, ILD interstitial lung disease
To identify patients with chronic fibrosing ILD with a progressive phenotype in the absence of specific diagnosis or procedure codes denoting progression, an algorithm was developed in consultation with expert pulmonologists based on their clinical experience using proxies for progression based on plausible markers of progression [12]. Patients were considered to have a progressive phenotype if they had any of the proxies for disease progression on or after the date of their second lung fibrosis code (Fig. 1). The index date for the cohort with chronic fibrosing ILD with a progressive phenotype was defined as the date a patient fulfilled the criteria for progressive disease. The proxies for progression were: ≥ 2 pulmonary function tests or ≥ 2 oxygen titration tests within 90 days; ≥ 2 high-resolution computed tomography (HRCT) or ≥ 3 chest computed tomography scans within 360 days; respiratory hospitalization; palliative care; lung transplant; any use of oxygen therapy or a corticosteroid > 20 mg (formulations with strength > 20 mg, not based on calculation or conversion); or new use of immunosuppressive therapy.
Patients with prevalent fibrosing ILD (i.e., who had two claims for fibrosing ILD during the baseline period and were excluded from the incident fibrosing ILD cohort) could be included in the cohort with incident chronic fibrosing ILD with a progressive phenotype if they had a proxy for progression on or after the date of the prevalent fibrosing ILD claim, without any of the proxies for progression during the baseline period.
Data are presented for patients with incident fibrosing ILD and separately for patients with fibrosing ILD with a progressive phenotype.
Outcomes
Patient demographics on the index date, medications received during the 12-month baseline period, and medications during the 12-month follow-up period were reported (see Fig. 1 for definitions of baseline and follow-up periods). Selected comorbidities during the baseline period were reported, and the Gagne combined comorbidity score (a single score calculated based on the presence or absence of a possible 20 comorbid conditions from the Charlson and Elixhauser comorbidity scores, with weight assigned according to associated mortality risk [14]) was calculated. Incident medical conditions were defined as claims during follow-up in patients who did not have claims for the event during the baseline period.
All-cause HCRU and costs were summed during the 12-month baseline period, excluding the 3 months prior to the index date (diagnosis of fibrosing ILD or identification of a progressive phenotype), and during the 12-month follow-up period (among the subset of patients with sufficient continuous enrollment). The 3 months prior to diagnosis were excluded to omit healthcare resource use associated with the diagnostic workups. Three months was the chosen duration as it is common for patients to attend the clinic every 3 months, and it may take this long to get all of the tests needed to confirm diagnosis. A similar approach has been taken in another recently published study in patients with IPF [15].
HCRU included claims for outpatient visits, hospitalizations, medications, critical care, emergency department visits and diagnostic tests. Total overall, inpatient, outpatient, emergency department, pharmacy and diagnostic test costs were estimated and adjusted for inflation to 2018 US dollars using the Consumer Price Index. Patients with capitated insurance plans were excluded from the cost analysis because payment in this arrangement is not tied directly to individual services provided. Costs were calculated using the total gross covered payment, which includes deductible, co-payment, co-insurance, coordination of benefits and net remaining paid by insurer. Total cost included all inpatient, outpatient, emergency department and pharmacy costs.
Inpatient mortality (diagnostic claim of sudden cardiac death or discharge status of death or expired) and incident medical conditions were reported for the whole cohort (including patients who had < 12 months follow-up). Mortality and incident medical condition data were not limited to the 12-month follow-up period. Follow-up was censored at outcome, end of data, disenrollment, or calendar date of 30 September 2015 (when ICD-9 coding in the US ended), whichever came first.
Statistical Methods
All analyses were descriptive and were conducted via the Aetion Evidence Platform® (version r3.11). Mean HCRU during baseline and follow-up periods was calculated as the number of claims in the baseline period (excluding 3 months prior to diagnosis) or follow-up period divided by the total number of eligible patients in each cohort with the required follow-up time. Mean medical costs during baseline and follow-up were calculated as the total costs incurred during baseline (excluding 3 months prior to diagnosis) or follow-up multiplied according to the 2018 inflation rate and divided by the number of eligible patients in each cohort with the required follow-up time. To account for the different durations of the baseline and follow-up periods, HCRU and costs for the 9-month baseline period were annualized by calculating the total divided by 275 days, multiplied by 365 days.
Data were described as mean (standard deviation [SD]) and median (interquartile range [IQR]) for continuous variables and n (%) for categorical variables. Incidence of medical conditions during follow-up was reported as the rate per 1000 person-years.
Results
Patients
Among 128,231 patients with a lung fibrosis claim, 23,577 (18%) had a second claim required for inclusion. Of these, 13,518 patients had incident progressive fibrosing ILD. A further 1204 patients with existing fibrosing ILD had proxies for progression, so the total in the incident chronic fibrosing ILD with a progressive phenotype cohort was 14,722 patients (Fig. 2a). In total, 9986 patients with incident fibrosing ILD, and 5840 with a progressive phenotype, had data available for 12-month follow-up (Fig. 2b).

a Patients included in the incident fibrosing ILD and chronic fibrosing ILD with progressive phenotype cohorts; b patients included in the baseline and 12-month follow-up cohorts and cost analysis. ILD interstitial lung disease, PF-ILD progressive fibrosing ILD
There were approximately equal proportions of males and females (Table 1). The mean (± SD) age was 68 (± 14) years in fibrosing ILD patients overall and 69 (± 14) years in those with a progressive phenotype; approximately 60% of patients were over 65 years of age. Patient employment status and health insurance plan details are shown in Table 1.
The most frequent comorbidities in the baseline period were chronic obstructive pulmonary disease (COPD) (44.8% of fibrosing ILD and 53.4% of those with a progressive phenotype) and type 2 diabetes mellitus (24.6% and 27.1%) (Supplementary Table 2). The most common incident medical conditions during follow-up were COPD (248.0 and 303.5 per 1000 patient-years in fibrosing ILD and in chronic fibrosing ILD with a progressive phenotype, respectively), arterial hypertension (182.23 and 223.34), and gastroesophageal reflux (181.07 and 190.05) (Supplementary Table 3).
The mean (± SD) Gagne combined comorbidity score [14] at baseline was 2.78 (± 2.92) in the fibrosing ILD cohort and 3.33 (± 3.03) in the cohort with a progressive phenotype (higher scores indicate a higher burden of comorbidities). The percentage of patients at baseline with a combined comorbidity score of 0 or 1 was 25.1% and 16.6% in the fibrosing ILD cohort and 18.6% and 14.7% in the cohort with a progressive phenotype.
Medications
The most frequently filled medications related to ILD were corticosteroids; 49.4% of the fibrosing ILD cohort and 56.6% of the cohort with a progressive phenotype received these drugs during baseline. In the 12-month follow-up period, 47.6% and 53.9% of patients received corticosteroids (Table 2). Proton pump inhibitors (not including those purchased over the counter) were common; at baseline they were filled by 38.7% of patients with fibrosing ILD and 42.4% of patients with a progressive phenotype and in the 12-month follow-up 41.3% and 45.1% of patients, respectively (Table 2).
Healthcare Resource Utilization
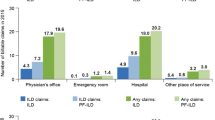
For HCRU and costs, both the overall data collected over the 9-month baseline period and the annualized data calculated from the 9 months are presented in Tables 3 and 4; here we describe the annualized results.
The annualized mean (± SD) number of claims from outpatient visits (including facility and physician service claims) during the baseline period was 30.0 (± 26.4) in the fibrosing ILD cohort and 34.1 (± 27.7) in the cohort with a progressive phenotype (Table 3). The mean number of outpatient visit claims in the 12-month follow-up period (among patients with sufficient follow-up) was 36.2 (± 28.6) in patients with fibrosing ILD and 41.9 (± 30.2) in the subgroup with a progressive phenotype (Table 3). The mean (± SD) number of claims for all-cause hospitalizations was 0.5 (± 1.1) during the baseline period and 0.6 (± 1.1) during 12-month follow-up in fibrosing ILD and 0.7 (± 1.2) during baseline and 0.7 (± 1.2) during 12-month follow-up in patients with a progressive phenotype.
Respiratory-related hospitalizations, intensive care unit admissions and emergency department visits in fibrosing ILD and in the subset with a progressive phenotype are shown in Table 3.
Healthcare Resource-Related Costs
Annualized mean (± SD) total costs during baseline were $40,907 (± 92,496) in the fibrosing ILD group and $49,561 (± 98,647) in patients with a progressive phenotype. In patients with 12 months of follow-up, mean costs were $46,157 (± 102,858) in the fibrosing ILD cohort and $54,215 (± 116,833) in the cohort with a progressive phenotype (Table 4). During baseline, annualized mean (± SD) outpatient costs were $19,088 (± 45,714) in fibrosing ILD and $22,663 (± 50,419) in patients with a progressive phenotype; outpatient and inpatient costs are shown in Table 4. Pharmacy costs and those related to fibrosing ILD/chronic fibrosing ILD with a progressive phenotype and selected comorbidities are also shown in Table 4.
During baseline, annualized mean diagnostic test costs in fibrosing ILD overall and in those with a progressive phenotype were $12,629 (± 62,056) and $16,485 (± 64,353), respectively; during follow-up, diagnostic test costs were $14,247 (± 69,370) and $17,863 (± 79,520), respectively (Table 4). The most frequent tests before and after the diagnosis of fibrosing ILD and chronic fibrosing ILD with a progressive phenotype included chest X-ray, pulmonary function tests and spirometry.
Inpatient Mortality
Inpatient mortality was 53.50 per 1000 patient-years (95% confidence interval [CI] 50.51, 56.50) over a median (IQR) of 295 (126–548) days of follow-up in the fibrosing ILD cohort and 77.44 per 1000 patient-years (72.75–82.13) over a median (IQR) of 270 (112–523) days of follow-up in the cohort with chronic fibrosing ILD with a progressive phenotype (Supplementary Table 4).
Discussion
This is one of the first claims-based studies to describe HCRU in patients with fibrosing ILD and chronic fibrosing ILD with a progressive phenotype. In this cohort of patients with fibrosing ILD, healthcare resource use was high. Healthcare resource use was higher in the subset of patients with a progressive phenotype than in the overall cohort. The high outpatient costs and high number of outpatient visit claims in chronic fibrosing ILD with a progressive phenotype likely reflect the careful monitoring of patients that is required in fibrosing ILD, which involves checking symptoms, exercise capacity, pulmonary function tests and HRCT. More than a third of patients were hospitalized during follow-up, and the median duration of hospitalization was 6–8 days (mean duration was 9–13 days), and therefore they incurred relatively high costs. Diagnostic test costs were also high in both cohorts; this is perhaps unsurprising given the challenges associated with the diagnosis of ILDs; a multidisciplinary approach is required [10], and HRCT imaging is considered a key diagnostic tool in detecting lung fibrosis [16]. Although we do not have data breaking down the cost of each type of test, it is likely that chest X-rays and HRCT were driving the diagnostic test costs since they are more costly than spirometry. Since we excluded the 3 months prior to diagnosis in the baseline data, our estimates do not include the diagnostic workups performed to make the initial fibrosing ILD diagnosis or identification of progression. However, it is possible that some diagnostic workup took place > 3 months prior to the index date.
Although the subset of patients with a progressive phenotype was not statistically significant compared with the larger group of patients with fibrosing ILD, we observed an appreciable difference in cost, which is very informative even without assessment of statistical difference.
There are few studies looking at HCRU in patients with chronic fibrosing ILD with a progressive phenotype, and it can be difficult to compare between studies because different methodologies are used for identifying patients. In addition, no international consensus exists on the definition of fibrosing ILD with a progressive phenotype, which was part of the motivation for the current study. Our results are similar to those reported in another US claims-based study that used pulmonologist visit frequency as a proxy to identify a progressive fibrosing phenotype in patients with ILD. The mean annual medical costs were $77,666 in the previous study compared with $54,215 (during follow-up) in the present study [11]. Although we present inpatient mortality during follow-up, it should be noted that these data are limited due to the method of capture, and we do not have data on deaths that occurred outside of hospital or on cause of death. A recently published single-center clinical cohort study in France found that overall survival in 165 patients with chronic fibrosing ILDs with a progressive phenotype was 83% at 3 years and 72% at 5 years, and mortality was higher in patients with faster disease progression [17].
A recent analysis of the placebo arms of three clinical trials (INBUILD and INPULSIS-1 and -2) found that patients with chronic fibrosing ILDs with a progressive phenotype other than IPF had a very similar disease course to patients with IPF in terms of forced vital capacity decline and risk of mortality [18]. This is reflected in similar healthcare costs observed in IPF and other chronic fibrosing ILDs with a progressive phenotype. The mean all-cause costs in the 12-month follow-up period in patients with a progressive phenotype ($54,215) in our study were similar to the annual costs in patients with IPF in a 2011 US claims study ($59,379) [19]. Another US claims study of patients with IPF in 2008 found lower direct medical costs ($26,378) but acknowledged that the definitions used for identifying patients with IPF affected the cost estimates [20]. Inpatient mortality in the 2008 study in IPF was 52.6 deaths per 1000 patient-years [20], which is lower than what we report for chronic fibrosing ILD with a progressive phenotype. These studies provide further evidence to support the idea that the burden on healthcare systems in patients with chronic fibrosing ILDs with a progressive phenotype other than IPF may be similar to IPF [10].
A high percentage of the patients with incident fibrosing ILD and those with a progressive phenotype had filled a prescription for a medication related to the treatment of fibrosing ILD prior to diagnosis. It is not possible to confirm the indication for the prescription since the databases do not provide this information. It is not surprising that the treatment patterns in the subset with a progressive phenotype were similar to the total fibrosing ILD cohort, since some of the commonly prescribed medications for fibrosing ILD were included as part of the algorithm-defining progressive disease. Furthermore, many of the medications could also be used for treatment of underlying diseases (e.g., autoimmune disease). Patients may also have been prescribed medications before a confirmed diagnosis of ILD based on their symptoms or for a different diagnosis that was later amended to progressive fibrosing ILD.
There was a high incidence of comorbidities in our analysis; similar to in IPF [21], cardiovascular conditions, COPD and gastroesophageal reflux were among the most common during follow-up. Approximately half of patients had prescription claims for treatment of comorbidities during baseline, though we do not know the intent of the prescriber. The combined comorbidity index showed a slightly higher rate of comorbidities in the cohort with a progressive phenotype than in the overall fibrosing ILD population. This should be considered when managing patients with fibrosing ILDs.
As there were no diagnosis codes for chronic fibrosing ILD with a progressive phenotype at the time of the study, we used a previously published algorithm [12]. However, some of these proxies may occur in the absence of progression, so we conducted a sensitivity analysis with a stricter definition of progression to ensure that single instances of oxygen therapy and respiratory hospitalization did not result in inclusion of patients that were not truly progressing. The stricter definition of progression had a few changes to the proxies: first, oxygen titration tests and use of corticosteroids were omitted as criteria for progression; second, respiratory hospitalization was retained, but only if there were two instances within 360 days, and oxygen therapy was retained only if there were two recorded claims during the study period. This led to minimal changes, and it was concluded that the potentially more sensitive proxies were not a major driver of the incidence and prevalence rates. It is possible that our estimates represent the upper limits of likely prevalence and incidence and include patients that might have been excluded if clinical data were available. Underlying conditions such as sarcoidosis are not always progressive; however, the results of the sensitivity analysis show no difference with stricter requirements for progression. Therefore, further analyses of the HCRU and costs using the stricter definition of progression are not presented here.
Our study has some limitations. There is a need to validate the algorithms used for chronic fibrosing ILD with a progressive phenotype for future research. Further, at the time of the initial exploration used to develop the algorithm, ICD-9 codes were used, as ICD-10 codes were not yet available in the data; this was due to the time lag between when a claim was generated in clinical practice and when it was fully adjudicated and available for research purposes. Although these findings are assumed to be generalizable to those of study cohorts generated using comparable ICD-10 codes, further exploration may be warranted, with a unique ICD-10 code specific to the progressive phenotype (introduced in 2021). The databases only include data from patients with commercial insurance and/or employer-sponsored Medicare supplemental insurance, so they represent a specific subset of the insured US population and may not be representative of Medicare patients overall. Claims databases provide real-world data of large cohorts; however, conclusions may be limited by the extent of information collected by the claims database itself (for example, smoking and weight data were not available) and the potential for time lag. Claims were filed for administrative purposes; ideally, conclusions would be based on clinical diagnosis and patient medical records. We did not investigate disease-related HCRU or costs, so we cannot say with certainty that healthcare visits and prescriptions were related to disease or progression. The study also included patients with a progressive phenotype in the overall fibrosing ILD cohort, meaning that it was not possible to compare outcomes in the populations with and without progressive disease. Since some of the outcomes measured are also included in the algorithm defining progression, there may be over-representation of certain procedures in the progressive phenotype subgroup. It will also be important for future studies to understand the added HCRU and cost burden associated with progressive disease compared with fibrosing ILD patients without progression.
Conclusions
Fibrosing ILD, either with or without a progressive phenotype, is associated with high HCRU and associated costs, and patients with a progressive phenotype have many outpatient claims and a high number of diagnostic tests. Further studies are needed to confirm these initial results and further investigate the burden of chronic fibrosing ILD with a progressive phenotype.
References
Antoniou KM, Margaritopoulos GA, Tomassetti S, Bonella F, Costabel U, Poletti V. Interstitial lung disease. Eur Respir Rev. 2014;23(131):40–54.
Kolb M, Vasakova M. The natural history of progressive fibrosing interstitial lung diseases. Respir Res. 2019;20(1):57.
Wijsenbeek M, Kreuter M, Olson A, et al. Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin. 2019;35(11):2015–24.
U.S. Food & Drug Administration. OFEV® (nintedanib): prescribing information. 2020 [cited 25 May 2021]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/205832s014lbl.pdf.
U.S. Food & Drug Administration. ESBRIET® (pirfenidone) prescribing information. 2019 [cited 12 March 2020]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/022535s012,208780s002lbl.pdf.
Richeldi L, Varone F, Bergna M, et al. Pharmacological management of progressive-fibrosing interstitial lung diseases: a review of the current evidence. Eur Respir Rev. 2018;27(150):180074.
De Sadeleer LJ, Goos T, Yserbyt J, Wuyts WA. Towards the essence of progressiveness: bringing progressive fibrosing interstitial lung disease (PF-ILD) to the next stage. J Clin Med. 2020;9(6):1722.
Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ, IPF Consensus Working Group. What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J. 2018;51(5):1800692.
Dieleman JL, Cao J, Chapin A, et al. US health care spending by payer and health condition, 1996–2016. JAMA. 2020;323(9):863–84.
Holtze C, Flaherty K, Kreuter M, et al. Healthcare utilisation and costs in the diagnosis and treatment of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180078.
Olson AL, Maher TM, Acciai V, et al. Healthcare resources utilization and costs of patients with non-IPF progressive fibrosing interstitial lung disease based on insurance claims in the USA. Adv Ther. 2020;37(7):3292–8.
Olson AL, Patnaik P, Hartmann N, Bohn RL, Garry EM, Wallace L. Prevalence and incidence of chronic fibrosing interstitial lung diseases with a progressive phenotype in the United States estimated in a large claims database analysis. Adv Ther. 2021;38(7):4100–14.
Centers for Disease Control and Prevention. International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM). 2015 [cited 9 October 2020]. https://www.cdc.gov/nchs/icd/icd9cm.htm.
Gagne JJ, Glynn RJ, Avorn J, Levin R, Schneeweiss S. A combined comorbidity score predicted mortality in elderly patients better than existing scores. J Clin Epidemiol. 2011;64(7):749–59.
Farrand E, Iribarren C, Vittinghoff E, et al. Impact of idiopathic pulmonary fibrosis on longitudinal health-care utilization in a community-based cohort of patients. Chest. 2021;159(1):219–27.
Walsh SLF, Devaraj A, Enghelmayer JI, et al. Role of imaging in progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180073.
Nasser M, Larrieu S, Si-Mohamed S, et al. Progressive fibrosing interstitial lung disease: a clinical cohort (the PROGRESS study). Eur Respir J. 2021;57(2):2002718.
Brown KK, Martinez FJ, Walsh SLF, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J. 2020;55(6):2000085.
Raimundo K, Chang E, Broder MS, Alexander K, Zazzali J, Swigris JJ. Clinical and economic burden of idiopathic pulmonary fibrosis: a retrospective cohort study. BMC Pulm Med. 2016;16:2.
Collard HR, Ward AJ, Lanes S, Cortney Hayflinger D, Rosenberg DM, Hunsche E. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ. 2012;15(5):829–35.
Mortimer K, Hartmann N, Chan C, Norman H, Wallace L, Enger C. Characterizing idiopathic pulmonary fibrosis patients using US medicare-advantage health plan claims data. BMC Pulm Med. 2019;19(1):11.
Acknowledgements
We wish to thank Julia A. Pisc and Jocelyn R. Wang of Aetion, Inc., contracted by Boehringer Ingelheim, for their contributions to the analytic implementation.
Funding
Sponsorship for this study (and Rapid Service Fee) was funded by Boehringer Ingelheim International GmbH.
Medical Writing and/or Editorial Assistance
Medical writing support was provided by Claire Scofield, M.Res., of MediTech Media, which was contracted and funded by Boehringer Ingelheim International GmbH, Ingelheim am Rhein, Germany. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
ALO, NH, RLB, DS and LW contributed to the study conception and design. Material preparation, data collection and analysis were performed by PP, EMG and MB. All authors contributed to the drafting and revision of the manuscript and provided approval to submit.
Prior Presentation
This manuscript is based on work that has been published as an abstract in Value in Health as part of the ISPOR, May 2020 supplement, but was not presented: https://doi.org/10.1016/j.jval.2020.04.1347.
Disclosures
Amy L. Olson has participated in speakers’ bureaus for Genentech and Boehringer Ingelheim, received fees from Boehringer Ingelheim and acted as Medical Advisor for MCG Diagnostics. Amy L. Olson changed affiliation during the completion of the manuscript; their current affiliation is: Boehringer Ingelheim Pharmaceuticals Inc., Ridgefield, CT, USA. Nadine Hartmann, Padmaja Patnaik, Michael Baldwin and Laura Wallace are employees of Boehringer Ingelheim. David Singer was an employee of Boehringer Ingelheim at the time of the present work; their current affiliation is: GlaxoSmithKline, Philadelphia, PA, USA. Elizabeth M. Garry is an employee of Aetion, Inc., in which she holds stock options. Aetion was contracted by Boehringer Ingelheim to execute the analyses used to generate the results of this study, but EMG received no direct payment. Rhonda L. Bohn has nothing to disclose.
Compliance with Ethics Guidelines
The use of the de-identified data was approved for exemption by the New England Independent Review Board (NEIRB).
Data Availability
The data that support the findings of this study are available from IBM Watson Health but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of IBM Watson Health.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Olson, A.L., Hartmann, N., Patnaik, P. et al. Healthcare Resource Utilization and Related Costs in Chronic Fibrosing Interstitial Lung Diseases with a Progressive Phenotype: A US Claims Database Analysis. Adv Ther 39, 1794–1809 (2022). https://doi.org/10.1007/s12325-022-02066-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-022-02066-9