
Abstract
Patients with myeloproliferative neoplasms (MPNs), a group of rare haematological conditions including polycythaemia vera, essential thrombocythaemia, and myelofibrosis, often experience a range of symptoms which can significantly impact their quality of life (QoL). Although symptom burden is highest in myelofibrosis and high-risk patients, lower-risk patients also report symptoms impacting their daily life and ability to work. In addition to physical symptoms, MPNs affect emotional well-being, with anxiety and depression frequently reported by patients. Despite significant advances in treatment options, such as the introduction of JAK1/JAK2 inhibitors, therapy for MPNs is often palliative; therefore, reduction of symptoms and improvement of QoL should be considered as major treatment goals. One of the main issues impacting MPN treatment is the discord between patient and physician perceptions of symptom burden, treatment goals, and expectations. New technologies, such as app-based reporting, can aid this communication, but are still not widely implemented. Additionally, regional variation further affects the psychosocial burden of MPNs on patients and their associates, as treatments and access to clinical trials are options for patients living in some areas, but not others. Overcoming some of the challenges in patient–physician communication and treatment access are key to improving disease management and QoL, as well as giving the patient greater input in treatment decisions.
Plain Language Summary
Myeloproliferative neoplasms (MPNs) are a group of blood diseases where the body makes too many blood cells. Patients with MPNs can have symptoms which interfere with their daily lives, such as tiredness, pain, sweating at night, dizziness, itching, and difficulty sleeping. They also often suffer from anxiety and/or depression. In nearly all cases, physicians cannot cure the disease, but drugs can prevent blood clots and reduce the speed at which the disease gets worse. Usually, the main aim of treatment is improving patients’ quality of life (QoL). Targeted drugs, such as ruxolitinib, treat MPNs and reduce symptoms, but do not cure the disease. Patients frequently want to play a bigger part in decisions about their treatment. However, physicians and patients often have different views on how well treatments are working and what to expect from the treatment. This can mean that patients feel they are not getting the best treatment for their symptoms. Also, patients may not be able to get some treatments or take part in a trial of a new drug, depending on where they live. This creates feelings of unfairness which can affect their mental health. Addressing all these problems may help improve the QoL for patients with these blood diseases.
Similar content being viewed by others


Why carry out this review? |
There is a key need in understanding the challenges presented in patient–physician communication and treatment access, to help improve disease management and quality of life in patients with myeloproliferative neoplasms (MPNs). |
What was learned from the review? |
One of the main issues impacting MPN treatment can be the conflict between patient and physician perceptions of symptom burden, treatment goals, and expectations. |
New technologies, such as app-based reporting, can help in improving patient–physician communication, but are still not widely implemented. |
The psychosocial burden of MPNs on patients and their associates can also be affected by regional variation, as treatments and access to clinical trials are options for patients living in some areas, but not others. |
What your patient wants you to know about living with myeloproliferative neoplasms |
Physical symptoms: |
Extreme fatigue - this feels like they cannot get out of bed for days on end. |
Bone pain - this can be excruciating. |
Pruritus - this severe itching interferes with daily life. |
Psychological and social impact: |
Burden of living with the knowledge that polycythaemia vera or essential thrombocythaemia could transform to myelofibrosis and progress to acute myeloid leukaemia - this creates anxiety which impacts all aspects of everyday life. |
Anxiety about what living with an incurable cancer will be like, including thoughts such as “how can I continue to live?”, “will I be in pain?”, and “what is the rest of my life going to be like?” |
Anxiety about impact on family; for example, how to continue providing for the family and how the family will survive after the patient’s death. |
Introduction
The myeloproliferative neoplasms (MPNs) polycythaemia vera (PV), essential thrombocythaemia (ET), and primary myelofibrosis (PMF) form a group of rare haematological malignancies with common clinical and pathogenic features [1]. PV, characterized by erythrocytosis, and ET, characterized by thrombocytosis, are both associated with an increased risk of major cardiovascular events and haemorrhage, and evolution to myelofibrosis (MF) (secondary or post-ET/post-PV MF), acute myeloid leukaemia, and, less commonly, myelodysplasia [2]. Such events are important causes of morbidity and mortality in patients with MPNs; overall, patients with primary or secondary MF, PV, or ET have increased risk of mortality compared with the general population [3]. Nevertheless, MPNs are chronic conditions for most patients, with median survivals of approximately 33 years for ET, 24 years for PV, and 15 years for PMF in younger (< 60-year-old) patients [4].
Patients with MPNs experience a broad array of symptoms, which overlap across the spectrum of PV, ET, and MF [1, 5]. These include microvascular-related symptoms, such as headache, vertigo, tinnitus, and dizziness, systemic manifestations (fatigue, night sweats, insomnia, body weight loss, and fever), pruritus, and splenomegaly, with associated abdominal pain and discomfort and early satiety [1, 5].
The overall burden of disease associated with MPNs has a significant negative impact on patients’ lives. Recent studies have shown that MPNs result in marked impairment of quality of life (QoL), work productivity, everyday life, and relationships [3, 6,7,8,9,10], and they are associated with significant emotional issues, including depression and anxiety [11, 12]. While the impact is generally greatest in patients with MF, and in higher-risk patients or those with greater symptom burden, negative effects are seen across the disease spectrum [3, 6, 7, 9].
In recent years, there have been major advances in our understanding of the underlying genetic changes which drive the development of MPNs, particularly with the discovery of mutations in the Janus kinase 2 (JAK2) [13], thrombopoietin receptor (MPL) [14], and calreticulin (CALR) [15] genes. These three mutations lead to constitutive activation of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signalling pathway, which is important for driving the production of blood cells from haematopoietic stem cells [16]. Which of these mutations is present influences both the characteristics and presentation of MPNs, as well as the prognosis [4, 17, 18]. Of these three key mutations, the JAK2 mutation, detected in 97% of patients with PV, 56% of patients with ET, and 50% of patients with PMF [13], has been widely targeted for drug development. Comprehensive molecular testing by next-generation sequencing has led to discovery of novel mutations which could become potential therapeutic targets [19, 20], and models based on identified associated genes provide risk classification based on their genomic profile [21, 22]. Ruxolitinib, a JAK1/JAK2 inhibitor, is the first such agent to be approved in MPNs, and represents a shift in the treatment paradigm from largely managing lifestyle and cardiovascular risk factors and addressing the effects of the disease, to attempting modification of the disease process itself. Despite these advances, nearly all treatment options for MPNs are not curative. Therefore, reduction of symptom burden, and thereby the impact of MPNs on QoL, is critical, and should be considered a major goal of treatment [1].
Recent treatment guidelines reflect current advances in diagnosis and treatment, and are largely, but not completely, in agreement regarding strategies. However, there is evidence of some differences in the way MPNs are diagnosed and managed between treatment settings and physician speciality, as well as between different countries [2, 23,24,25]. As in other disease settings, there is also often marked discord between physician and patient perception around issues such as patients’ understanding of their disease, its management and treatment goals, and of the disease burden experienced [26].
This review intends to give a brief overview of the current MPN treatment options, and provide an assessment from the viewpoint of patient advocates, with the aim of highlighting issues around MPNs and patient needs. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Treating MPNs
In recent years, a number of national and international guidelines for the diagnosis and treatment of patients with MPNs have been published or updated to reflect advances in the understanding of the disease and the availability of new treatment options (Table 1) [27,28,29,30,31,32,33,34]. The discovery of the three driver mutations has also led to mutation screening currently being recommended as part of the diagnostic criteria of MPNs [27,28,29,30,31,32, 34]. The guidelines provide similar advice with regard to treatment, although, in some cases, they reflect availability and payment status of particular drugs, or local practice. Treatment options are recommended on the basis of a combination of the underlying diagnosis (PV, ET, or MF) and prognostic risk. For MF, scoring systems such as the International Prognostic Scoring System (IPSS) are used at diagnosis, and “dynamic” IPSS (DIPSS) or DIPSS-plus for assessment during treatment [27,28,29, 31, 32]. Patients with PV and ET are generally stratified as high or low risk on the basis of age and thrombosis history, with some guidelines recommending further classification of patients with PV using the revised International Prognosis Score of Thrombosis for ET (IPSET thrombosis), which also takes into account cardiovascular risk factors and JAK2 mutation status [35].
Currently, the only curative approach for MPNs is allogeneic haematopoietic stem cell transplantation (ASCT), but given that it carries a considerable risk of mortality and morbidity, it is generally reserved for younger patients with MF with higher prognostic risk disease. For other patients, the majority of conventional therapies for MPN are largely palliative (Table 1). In PV and ET, these are predominantly aimed at reducing the risk of occurrence or reoccurrence of thrombosis, and ultimately reducing the risk of progression to MF; and for patients with MF, who are not suitable for ASCT, treatment is generally guided by the predominant symptoms, mainly anaemia and splenomegaly.
Targeted Therapies
The most important advance in treatment of MPNs in the last decade came with the development of the JAK1/JAK2 inhibitors. The JAK1/JAK2 inhibitor ruxolitinib was the first targeted therapy to be widely approved globally for MPN [36]; and more recently, the JAK2 inhibitor fedratinib has been approved by the US Food and Drug Administration (FDA) for intermediate-2 or high-risk primary or secondary MF [37]. In clinical trials, ruxolitinib was shown to significantly reduce spleen size, ameliorate debilitating MF-related symptoms, and improve QoL in patients with intermediate-2 or high-risk MF versus best available treatment, with overall survival also improved versus placebo [38, 39]. The benefits of ruxolitinib on spleen size and overall survival have been shown to be maintained over longer-term therapy [40, 41], and sustained treatment was also associated with greater odds of bone marrow fibrosis improvement or stabilization and decreased odds of bone marrow fibrosis worsening in a large proportion of patients with MF versus best available treatment [42]. Additionally, ruxolitinib has demonstrated significant reductions in spleen size, control of haematocrit, and improvement of PV-related symptoms. Improved QoL was also seen compared with standard therapy in patients with PV and splenomegaly and an inadequate response to or intolerance of hydroxyurea [43, 44]. In addition, durable improvements in haematocrit and symptoms in patients without splenomegaly have been observed [45]. On the basis of these trials, ruxolitinib has been widely approved for the treatment of splenomegaly or symptoms in patients with MF (PMF or MF secondary to PV or ET) and for patients with PV who are resistant to or intolerant of hydroxyurea, and it is recommended, generally, as a second-line treatment option, in most treatment guidelines for PV (Table 1).
While the treatment options have been significantly improved with the introduction of ruxolitinib, JAK1 and JAK2 inhibitors have been associated with immunosuppression by targeting reducing type 1 and 2 cytokine production, and modulating dendritic cell function [46, 47]. This immunosuppression can result in increased risk of infections and reactivation of existing infections, such as herpes zoster. Ruxolinitib has also been associated with an increased risk of aggressive B cell lymphoma, although further studies are needed to establish whether JAK1 and/or JAK2 inhibition plays a role in this increased risk [48]. Additionally, not all patients are candidates for treatment, and, although ruxolitinib is generally well tolerated, some patients do experience issues which can lead to dose reduction and treatment discontinuation. There is also a population of patients who respond initially to ruxolitinib but in whom treatment subsequently fails, with re-emergence of splenomegaly and constitutional symptoms [49]. In addition, although it provides substantial clinical benefit, ruxolitinib treatment is not curative in terms of complete haematological remission with normalization of blood counts or reduction in the mutant allele burden, and transformation to acute leukaemia can still occur [16]. A number of studies are looking at the potential for combinations of ruxolitinib with traditional and experimental agents, with the aim of, for example, maintaining platelet count or reducing anaemia, to maximize ruxolitinib dose, or to improve outcome (e.g., better spleen response, fibrosis improvement, greater reduction in mutant allele burden) [5, 50]. While this approach has shown potential for some combinations, larger studies and validated criteria for judging success are needed.
Clinical trials of the JAK2 inhibitor fedratinib in both JAK-naïve patients with MF and, importantly, those previously treated with ruxolitinib showed a clinical benefit of treatment in both patient groups [51, 52]. Overall, just over a third of JAK-naïve and half of patients previously treated with ruxolitinib achieved at least a 35% reduction in spleen volume. In addition, 40% of patients experienced at least a 50% reduction in MF-related symptoms. However, trials were terminated following a possible association with suspected cases of Wernicke's encephalopathy, a neurological emergency resulting from thiamine (vitamin B1) deficiency and characterized by mental status changes, confusion and memory problems, and gait and oculomotor dysfunction [51]. The FDA subsequently placed fedratinib on clinical hold, pending investigation of the suspected link; however, this was removed following submission of further data by Celgene, and fedratinib was approved by the FDA for adults with intermediate-2 or high-risk primary or secondary (post-PV or post-ET) MF. The prescribing information for fedratinib includes a boxed warning to advise healthcare professionals and patients about the risk of serious and fatal encephalopathy, including Wernicke’s encephalopathy, and healthcare professionals are advised to assess thiamine levels in all patients prior to starting fedratinib, periodically during treatment, and as clinically indicated. If encephalopathy is suspected, fedratinib should be immediately discontinued and parenteral thiamine initiated [37]. At the time of writing, the position of fedratinib in treatment algorithms and recommendations has yet to be confirmed in treatment guidelines; however, it clearly offers an encouraging option for those patients who have failed therapy with ruxolitinib.
New Agents
A number of other JAK inhibitors have been developed, and although some have entered clinical trials in patients with MPN, their development has been delayed by early toxicity issues, or a lack of benefit over ruxolitinib. Firstly, momelotinib, a JAK1/JAK2 inhibitor, showed similar responses to ruxolitinib with respect to reduction in spleen size, but lesser alleviation of constitutional symptoms in phase 3 trials in patients with MF, although momelotinib was associated with a reduced transfusion requirement relative to ruxolitinib [53, 54]. Long-term data failed to show a survival benefit of treatment with momelotinib, and demonstrated a high incidence of peripheral neuropathy [55], and studies of momelotinib in PV and ET were discontinued as a result of limited efficacy [56]. However, the FDA has recently granted fast-track designation to momelotinib for the treatment of patients with intermediate- or high-risk MF who previously received a JAK inhibitor, and a phase 3 trial to evaluate the activity of momelotinib for symptomatic, anaemic patients with MF previously treated with JAK inhibitor therapy is planned [57].
The JAK2/FMS-like tyrosine kinase 3 (FLT3) inhibitor pacritinib achieved significantly greater reductions in spleen size and improvements in symptoms versus best available therapy, including ruxolitinib, in patients with MF and thrombocytopenia [58]. However, pacritinib was placed on clinical hold by the FDA in 2016, following interim overall survival results from PERSIST-2 which showed a detrimental effect on survival consistent with the results from PERSIST-1 [59]. This hold was removed following submission of the final study reports for PERSIST-1 and -2 trials, and a commitment to undertake a dose-exploration clinical trial (NCT03165734; available from https://clinicaltrials.gov/ct2/show/NCT03165734) at the FDA’s request [60].
A number of emerging therapies beyond JAK inhibitors against a range of targets have been studied or are currently undergoing trials, but, to date, none are near to becoming available in the clinic [5, 50]. For example, a number of drugs targeting the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt)-strain transforming/rapamycin kinase pathway are in early development or have completed phase 1 or phase 2 trials (e.g., everolimus, buparlisib). Histone deacetylase inhibitors are also being assessed for clinical efficacy in MPN, with panobinostat, vorinostat, and pracinostat having completed phase 2 studies. Other therapies undergoing early phase clinical trials include telomerase inhibitors (e.g., imetelstat), Hedgehog pathway inhibitors, and antifibrotic agents (e.g., PRM-151). Additional emerging potential therapies include CRISPR-Cas9 and immunotherapy, but these are not yet ready to enter clinical testing [5].
Impact of MPNs on Patient-Reported Outcomes (Pros)
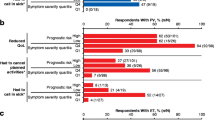
The wide-ranging nature and degree of the impact of MPNs has been highlighted in an increasing number of studies over recent years [3, 6,7,8,9,10,11,12]. One of the largest studies was the international MPN Landmark survey, conducted from April 2016 to October 2016, which included data from over 800 patients with MPN and over 450 physicians across the USA, and almost 700 patients with MPN and over 200 physicians across Australia, Canada, Germany, Italy, Japan, and the UK [3, 7]. The results confirmed the broad symptom burden experienced by patients with MPN across the disease spectrum, with the vast majority of patients (90%) entering the survey having experienced MPN-related symptoms over the previous 12 months. In common with other patient surveys, the most frequently reported symptom among all MPN subtypes was fatigue (Fig. 1) [6]. Other commonly reported symptoms varied depending on disease subtype: abdominal discomfort, night sweats, difficulty sleeping, and bone pain were most common in patients with MF; pruritus, concentration problems, and night sweats in patients with PV, and dizziness, night sweats, bruising, itching, and difficulty sleeping in patients with ET. Most patients reported that their MPN-related symptoms affected their QoL (MF, 81%; PV, 66%; ET, 57% in the US cohort; MF, 83%; PV, 72%; ET, 74% in the rest of the world cohort), with many patients also reporting an impact on their ability to work, and on relationships and family life. A common and important finding in the survey was that, although high-prognostic risk patients and those with the most severe symptom burden were more likely to report that their disease had an impact on QoL, daily living, and work ability, such issues were still reported by a significant proportion of lower-risk patients and those with lower symptom burden (Fig. 2).

a Reproduced from Mesa et al. [3]. BMC Cancer. 2016 Feb 27;16:167 © 2016, Mesa et al. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), and b Modified from Harrison et al. [7] Ann Hematol. 2017;96:1653–65 © 2017, Harrison et al. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), bars made vertical
Symptoms experienced by patients with MPN in the Landmark Survey in a US cohort and b rest of the world cohort. ET essential thrombocythaemia, MF myelofibrosis, MPN myeloproliferative neoplasm, PV polycythaemia vera.

Reproduced from Mesa et al. BMC Cancer. 2016 Feb 27;16:167 © 2016, Mesa et al. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), no changes
Impact of MPNs on QoL, work, and activities of daily living. MPN impact was stratified by calculated prognostic risk score and symptom severity quartile in respondents with a MF, b PV, and c ET. ET essential thrombocythaemia, MF myelofibrosis, MPN myeloproliferative neoplasm, PV polycythaemia vera, Q1 quartile 1, Q4 quartile 4, QoL quality of life. a ≥ 1 day in the preceding 30 days.
The findings from the Landmark study are echoed in other surveys of PROs in MPN [6, 8,9,10]. In a study conducted in the UK and USA, significant reductions in QoL relative to controls were observed, as well as differences in QoL between UK and US patients, and in symptom burden between male and female patients [6]. This last observation was also reported in a study by the MPN QoL International Working Group [61]. The US “Living with MPNs” survey found that all three types of MPNs have a substantial negative impact on patients’ employment status, career potential, and work productivity comparable to the impairment levels reported for other disabling chronic conditions [9, 10]. While patients with MF and those with higher symptom burden reported the greatest impact, a notable proportion of patients with PV and ET and lower symptom burden were also severely impacted by their symptoms and corresponding impairments associated with their disease [9, 10].
In addition to the effect of MPN on overall QoL, working ability, and relationships, the Landmark survey and other recent studies have also highlighted the impact of MPN on patients’ emotional well-being. Negative impacts on mental well-being were common, with around a quarter of patients reporting they frequently felt anxious or worried; and over 60% reporting having felt some level of depression over the previous month, of whom 22% indicted that this depression had a high impact on them [7]. Higher comorbidity burden, total symptom burden, fatigue burden, lower functional level, and global health/QoL in patients with MPN have been shown to be associated with increased odds of anxiety and depression [11], with depression being uniquely associated with overall physical symptom burden [12].
Finally, as with many chronic diseases, MPN also leads to significant financial burden for many patients. In the USA, many patients are unable to get (full) insurance coverage for MPN medications, leading to substantial additional annual healthcare costs [62]. As well as the direct costs of treatments, hospital stays, and travel to and from physician appointments, reduced ability to work leads to increased financial burden in patients with MPN. In the MPN Landmark survey, substantial losses in household income were observed in patients with reduced work hours, discontinued employment, and medical disability [63]. Financial stress can further impact emotional well-being, with increased financial burden associated with increased risk anxiety and depression in patients with MPN [11].
The findings from all of these studies strongly suggest that addressing disease burden in patients with MPN as part of the overall management strategy is crucial to minimize impact on their daily lives and emotional and physical well-being. They also stress that, while guidelines generally recommend treatment for patients with higher prognostic risk, those with lower risk may also suffer from significant symptom-related morbidity impacting their QoL and day-to-day lives. Clear communication between the patient and physician is therefore essential if symptoms are to be recognized and managed. However, the Landmark study revealed that, similar to other oncology settings [64, 65], there is often a marked discord between physician and patient perceptions of MPN, its treatment and impact, and how well these issues are discussed and communicated [26]. Particularly striking differences were found in aspects such as management, treatment goals, and expectations, with marked discord between what patients and physicians reported having been discussed. For example, many patients in the Landmark study were unaware that some common symptoms such as difficulty sleeping, dizziness, vertigo, and light-headedness were MPN-related, whereas physicians reported that, in their opinion, patients were generally able to recognize symptoms as being disease-related. Overall, the survey found that physicians tended to under-report symptoms and under-recognize their impact on QoL compared with patient perceptions (Fig. 3).

Reproduced with permission from Mesa et al. [26] © 2016 The Authors. Cancer published by Wiley Periodicals, Inc. on behalf of American Cancer Society
Myeloproliferative neoplasm-related symptoms at diagnosis among a patient and b physician respondents. *The question to patient respondents was “Which of these symptoms were you experiencing at the time of diagnosis?” The analysis included the percentages of patient respondents who did not answer “none.” †The question to physician respondents was “Out of 100%, what proportion of all newly diagnosed patients do you estimate have no symptoms?” The analysis included the median value provided by physician respondents for the proportion of newly diagnosed patients with symptoms. ET essential thrombocythaemia, MF myelofibrosis, PV polycythaemia vera.
Such discord between patient and physician perceptions may lead to suboptimal disease management. In the Landmark study, for example, although most patients were treated in line with current guidelines, around a quarter of patients managed with watchful waiting had a moderate to high symptom burden, yet did not receive any drug therapy to address this [7]. Lessening of differences in perception surrounding prognosis, symptom control, and treatment goals between patients and physicians is clearly important to improving outcomes in patients with MPN. In general, patients want to be more involved with their journey, and share decisions regarding their treatment and management. Overall, there is a need for improvements in communication between physician and patient, and for better patient education. Additional scientific research on PROs and QoL measures may also increase interest in the use of these measures in improving disease management and patient QoL [1].
Unmet Needs of Patients with MPNs
Regionality
In the authors’ experience, as part of a global network of patient advocates, it is clear that in MPN, as in many other disease areas, management strategies and access to treatments are not standard globally. Limited access to the best/current therapy and management has the potential to significantly impact on patients’ QoL if this results in greater symptom burden. In addition, in the era of social media, patients seek out and share information via multiple sources, including national and international patient support and patient advocacy groups. Hence patients are made aware of treatments and new developments which, while available elsewhere, are not an option for them in their own country, engendering a feeling of a “second class” standard of care. Such a perception can lead to frustration and anxiety, and so impact on patients’ well-being in general.
Regional Variability in Access to MPN Treatment Options
As previously discussed, national and international guidelines are used to recommend treatment and management strategies. However, although many such guidelines are updated regularly, they are based on the data available in that snapshot of time, and may not reflect new advances or include recently approved treatment options. For example, although approved for use in MF in 2014, the most recent Japanese treatment guidelines published in 2017 do not include recommendations for the use of ruxolitinib, pending data from trials conducted in Japan and coverage by Japanese National Health Insurance [33].
Even despite guideline recommendations, the use of some recommended agents may be limited by availability and payment status of particular drugs, or local practice. For example, the use of some agents is limited as they are not covered by national health insurance, including anagrelide in Japan [33]. Similarly, although widely recommended as a cytoreductive option, interferon-α use in MPNs is off-label and hence patients (e.g. in the USA) may struggle to obtain insurance coverage, particularly for the pegylated form which is associated with fewer side effects [66].
Regional Variability in Access to Clinical Trials
Despite some evidence of a slight migration of clinical trial sites to low- and middle-income countries with emerging economies, the vast majority are conducted in high-income countries, predominantly the USA [67]. Data from clinicaltrials.gov shows that of over 315,000 privately and publicly funded clinical studies registered as of September 2019, 34% were located in the USA alone, with a further 5% located in both the USA and a non-US country [68]. Western Europe had the second largest number of registered trials (28.5%), with only approximately 3% of clinical trials conducted in South America and Africa, 1.5% in South Asia, and < 1% in Central America. While participation in clinical trials has been suggested as a means of improving access to medication in low- to middle-income countries, the ability to run such trials is often beset by problems, including regulatory issues, lack of infrastructure, and availability of trained clinical researchers [67, 69]. Nevertheless, to ensure robust data reflecting the whole MPN population, patient involvement from other countries should be facilitated, if possible, with consideration given to including centres outside of the USA and Europe for clinical trials in MPN and for international collaboration between clinicians in different countries.
Locating and engaging patients is critical for the success of clinical trials. Patients mostly welcome being asked to participate in research: in a survey of patients with cancer conducted by the UK National Health Service (NHS), only a third said that taking part in research had been discussed, but of these 95% said they were glad to have been asked, and over half of those with whom this had not been discussed said they would have liked to have been asked [70]. In addition to patient participation, trial design is also a major consideration. Currently, treatment guidelines are largely driven by data generated in randomized controlled trials (RCTs). However, while RCTs are the “gold standard” to determine whether a particular drug or intervention is effective, by necessity they are highly controlled and carried out in relatively small, tightly defined, highly selected patient samples, meaning that they may not fully reflect the patients seen by practicing physicians in their particular “real-world” setting [71]. Novel trial designs may provide more appropriate data, while also improving patient access; for example, pragmatic trials which are designed to show patient-relevant effectiveness of an intervention in broad patient groups, reflecting the population and management strategies in real-world practice [71]. Additionally, the use of technology, ranging from simple telephone contact, through web and smartphone tools and medical-grade wearable sensors to collect real-world data in home settings has the potential to reduce the costs, inconvenience, and labour associated with regular clinic attendance required during trials [72]. Such tactics may prove particularly beneficial for improving patient inclusion and robustness of data in rare diseases, such as MPN, where specialists are thinly spread and geographic proximity to trial centres is an issue.
Pros: Improving Real-World Evidence
Assessment of QoL is one of the only assessments for patients to monitor their disease, other than blood count measures. Given the importance of these PROs in incurable MPNs and despite an increase in interest and assessment of these measures in clinical trials, there is a general lack of data. A key challenge is the development of a consistent and quantitative method for capturing and measuring PROs. The Myeloproliferative Neoplasms Symptom Assessment Form (MPN-SAF) has been validated in patients with all forms of MPN, and is a comprehensive and reliable 27-item instrument which aims to concisely assesses the prevalence and severity of symptoms [73]. More widespread use of validated, widely approved, and standardized scoring systems has the potential to provide large amounts of real-world data, and also allow for more accurate comparisons between clinical trials.
App-based symptom trackers are a powerful way to collect patient data and to improve patient–physician communication about symptom severity and impact on QoL. Such technological tools allow patients to take ownership of assessment of their disease progress and provide a longitudinal measure of symptoms over time, which could additionally aid physicians in early recognition of disease progression. While potentially providing insightful data on the patient’s perspective and facilitating patient–physician communication, these sorts of tools have not yet been widely adopted, which limits their usefulness in a broad clinical setting. Additionally, data-protection restrictions make large-scale analyses of personal patient data challenging. Despite these limitations, these methods of data collection could aid in improving disease management for many patients when rolled out more universally.
Psychosocial Burden of Illness on Patients and Their Environment: Living from Day to Day with MPN
The classification of MPN as a “blood cancer” can be a double-edged sword as the thought of “cancer” as opposed to a “chronic disease” may have a negative impact in terms of how patients share this information with others, including family and employers, and may increase fear and anxiety in patients and their family and caregivers. On the other hand, it may have some benefits (e.g., gateway to access to certain facilities/help in some countries) and may make employers, for example, take the disease more seriously as more than just a chronic condition.
It is critical that physicians realize that understanding the impact of MPN on patients goes beyond an assessment of their symptoms. For example, the psychosocial burden of the disease on the patient and their associates (caregiver, spouse, children, siblings, friends, co-workers, employer, etc.) can be significant. In common with other chronic untreatable diseases, patients with MPN often live with a high level of anxiety and depression on a daily basis [7, 11, 12]. While this is attributable, at least in part, to symptom burden [11, 12], other factors play a part. Anxiety can arise from individual and diverse underlying issues. For example, patients feeling that they are not living up to the standards they set for themselves as a partner, parent, or worker; financial difficulties; and sexuality-related symptoms, which are often unaddressed by patients and physicians, and can impact on intimacy with partners [63]. Current scoring systems do not adequately reflect these burdens; for example, the MPN-SAF asks for a simple assessment of QoL, “sad mood” and depression, and problems with sexual desire or function on a scale of 0–10, but cannot assess the individual specific, complex, and multifactorial issues underlying these scores. There is, therefore, a need for tools which are more “wholistic”, which include multiple aspects of patients’ lives, including an assessment of impact on family and caregivers. If such tools became available, and even in their absence, the challenge is how to address the need for and provision of support to address such problems given that they are not universally acknowledged or available.
New Studies of Key Issues Around Unmet Needs
The MPN Advocates Network Global Patient Survey aims to assess unmet needs and identify the underserved MPN population. The survey was designed by MPN advocates in conjunction with their medical advisory committee. Distributed through a global network of country-specific MPN patient organizations, it aims to understand the unmet needs around the world with the aim of improving access to information, expertise, and new therapies. Following on from the findings of the first Landmark study, Landmark 2.0 aims to further define patients’ unmet needs that were initially identified; to further investigate the discord between physician and patient perceptions of symptoms, disease burden, and overall health; and to increase the number of countries included in the study population. While there are a number of similar surveys carried out at regional and national levels, the global nature of the Landmark studies allows assessment of regional variability which can pinpoint areas for future in-depth research.
Conclusion
Despite recent advances in MPN treatment, MPN remains a broadly incurable chronic condition with a large impact on patients’ daily lives. Large global studies have identified regional variability in the unmet needs of patients with MPN, with treatment accessibility, guidelines, and access to clinical trial participation depending on the region or country where the patient lives. While PROs are one of the key measures of patient QoL, real-world data are limited. Widespread implementation of technologies which allow standardization and quantification of PROs by patients and physicians can aid in joint decision-making for optimizing treatment, and give patients ownership of their disease management.
References
Abruzzese E, Niscola P, Trawinska MM, de Fabritiis P. Chronic myeloproliferative disorders: is quality-of-life the new goal? Curr Med Res Opin. 2018;34:1345–7.
Vannucchi AM, Guglielmelli P. What are the current treatment approaches for patients with polycythemia vera and essential thrombocythemia? Hematology Am Soc Hematol Educ Program. 2017;2017:480–8.
Mesa R, Miller CB, Thyne M, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer. 2016;16:167.
Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124:2507–13 (quiz 2615).
Vannucchi AM, Harrison CN. Emerging treatments for classical myeloproliferative neoplasms. Blood. 2017;129:693–703.
Anderson LA, James G, Duncombe AS, et al. Myeloproliferative neoplasm patient symptom burden and quality of life: evidence of significant impairment compared to controls. Am J Hematol. 2015;90:864–70.
Harrison CN, Koschmieder S, Foltz L, et al. The impact of myeloproliferative neoplasms (MPNs) on patient quality of life and productivity: results from the international MPN Landmark survey. Ann Hematol. 2017;96:1653–65.
Mesa R, Boccia RV, Grunwald MR, et al. Patient-reported outcomes data from REVEAL at the time of enrollment (baseline): a prospective observational study of patients with polycythemia vera in the United States. Clin Lymphoma Myeloma Leuk. 2018;18:590–6.
Yu J, Parasuraman S, Paranagama D, et al. Impact of myeloproliferative neoplasms on patients’ employment status and work productivity in the United States: results from the living with MPNs survey. BMC Cancer. 2018;18:420.
Yu J, Paranagama D, Geyer HL, Parasuraman S, Mesa R. Relationship between symptom burden and disability leave among patients with myeloproliferative neoplasms (MPNs): findings from the Living with MPN patient survey. Ann Hematol. 2019;98:1119–25.
Brochmann N, Flachs EM, Christensen AI, et al. Anxiety and depression in patients with Philadelphia-negative myeloproliferative neoplasms: a nationwide population-based survey in Denmark. Clin Epidemiol. 2018;11:23–33.
McFarland DC, Shaffer KM, Polizzi H, et al. Associations of physical and psychologic symptom burden in patients with Philadelphia chromosome-negative myeloproliferative neoplasms. Psychosomatics. 2018;59:472–80.
Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61.
Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270.
Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–405.
Greenfield G, McPherson S, Mills K, McMullin MF. The ruxolitinib effect: understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J Transl Med. 2018;16:360.
Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123:1544–51.
Spivak JL. Myeloproliferative neoplasms. N Engl J Med. 2017;376:2168–81.
Alduaji W, McNamara CJ, Schuh A, et al. Clinical utility of next-generation sequencing in the management of myeloproliferative neoplasms: a single-center experience. HemaSphere. 2018;2:3.
Chang YC, Lin HC, Chiang YH, et al. Targeted next-generation sequencing identified novel mutations in triple-negative myeloproliferative neoplasms. Med Oncol. 2017;34(5):83.
Grinfeld J, Nangalia J, Baxter EJ, et al. Disease heterogeneity and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416–30.
Marneth AE, Mullally A. The molecular genetics of myeloproliferative neoplasms. Cold Spring Harb Perspect Med. 2020;10(2):a034876.
Kaifie A, Isfort S, Gattermann N, et al. Health care setting and severity, symptom burden, and complications in patients with Philadelphia-negative myeloproliferative neoplasms (MPN): a comparison between university hospitals, community hospitals, and office-based physicians. Ann Hematol. 2016;95:1399–410.
Ellis MH, Koren-Michowitz M, Lavi N, Vannucchi AM, Mesa R, Harrison CN. Ruxolitinib for the management of myelofibrosis: results of an international physician survey. Leuk Res. 2017;61:6–9.
Habib LA, Kuo KHM, Panzarella T, Gupta V, Trinkaus M. Management of polycythemia vera: a survey of Canadian physician practice patterns. Clin Lymphoma Myeloma Leuk. 2019;19:e37–42.
Mesa RA, Miller CB, Thyne M, et al. Differences in treatment goals and perception of symptom burden between patients with myeloproliferative neoplasms (MPNs) and hematologists/oncologists in the United States: findings from the MPN Landmark survey. Cancer. 2017;123:449–58.
NCCN Clinical Practice Guidelines in Oncology. Myeloproliferative neoplasms. Version 2.2019–October 29, 2018. 2019. https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf. Accessed Aug 2019.
Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. 2018;32:1057–69.
Vannucchi AM, Barbui T, Cervantes F, et al. Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v85–99.
McMullin MF, Harrison CN, Ali S, et al. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. Br J Haematol. 2019;184:176–91.
Nordic MPN Study Group. Nordic guidelines on the diagnosis and treatment of patients with myeloproliferative neoplasms. 2019. https://nmpn.org/index.php/guidelines/17-nmpn-care-program-2017/file. Accessed Mar 2020.
Agarwal MB, Malhotra H, Chakrabarti P, et al. Revised myeloproliferative neoplasms working group consensus recommendations for diagnosis and management of primary myelofibrosis, polycythemia vera, and essential thrombocythemia. Indian J Med Paediatr Oncol. 2018;39:503–15.
Usui N. JSH guideline for tumors of hematopoietic and lymphoid tissues-lukemia: 4. Chronic myelogenous leukemia (CML)/myeloproliferative neoplasms (MPN). Int J Hematol. 2017;106:591–611.
Choi CW, Bang SM, Jang S, et al. Guidelines for the management of myeloproliferative neoplasms. Korean J Intern Med. 2015;30:771–88.
Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015;5:e369.
JAKAFI (ruxolitinib) [prescribing information] Wilmington, DE, Incyte Corporation. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/202192s017lbl.pdf. Accessed Sep 2019.
INREBIC (fedratinib) [prescribing information]. Summit, NJ, Celgene Corporation. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212327s000lbl.pdf. Accessed Sep 2019.
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807.
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–98.
Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3-year follow-up of COMFORT-I. Haematologica. 2015;100:479–88.
Verstovsek S, Gotlib J, Mesa RA, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol. 2017;10:156.
Kvasnicka HM, Thiele J, Bueso-Ramos CE, et al. Long-term effects of ruxolitinib versus best available therapy on bone marrow fibrosis in patients with myelofibrosis. J Hematol Oncol. 2018;11:42.
Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372:426–35.
Mesa R, Verstovsek S, Kiladjian JJ, et al. Changes in quality of life and disease-related symptoms in patients with polycythemia vera receiving ruxolitinib or standard therapy. Eur J Haematol. 2016;97:192–200.
Griesshammer M, Saydam G, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythemia vera without splenomegaly: 80-week follow-up from the RESPONSE-2 trial. Ann Hematol. 2018;97:1591–600.
Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib-associated infections: a systematic review and meta-analysis. Am J Hematol. 2018;93(3):339–47.
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O'Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;17(1):78.
Porpaczy E, Tripolt S, Hoelbl-Kovacic A, et al. Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy. Blood. 2018;132(7):694–706.
Pardanani A, Tefferi A. Definition and management of ruxolitinib treatment failure in myelofibrosis. Blood Cancer J. 2014;4:e268.
Bose P, Verstovsek S. JAK2 inhibitors for myeloproliferative neoplasms: what is next? Blood. 2017;130:115–25.
Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1:643–51.
Harrison CN, Schaap N, Vannucchi AM, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017;4:e317–e324.
Mesa RA, Kiladjian JJ, Catalano JV, et al. SIMPLIFY-1: a phase III randomized trial of momelotinib versus ruxolitinib in Janus kinase inhibitor-naïve patients with myelofibrosis. J Clin Oncol. 2017;35:3844–50.
Harrison CN, Vannucchi AM, Platzbecker U, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018;5:e73–81.
Tefferi A, Barraco D, Lasho TL, et al. Momelotinib therapy for myelofibrosis: a 7-year follow-up. Blood Cancer J. 2018;8:29.
Verstovsek S, Courby S, Griesshammer M, et al. A phase 2 study of momelotinib, a potent JAK1 and JAK2 inhibitor, in patients with polycythemia vera or essential thrombocythemia. Leuk Res. 2017;60:11–7.
Sierra Oncology. Sierra announces momelotinib granted FDA fast track designation. Press release. 2019. https://investor.sierraoncology.com/2019-06-05-Sierra-Announces-Momelotinib-Granted-FDA-Fast-Track-Designation. Accessed Sep 2019.
Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4:652–9.
Mascarenhas J, Hoffman R, Talpaz M, et al. Results of the persist-2 phase 3 study of pacritinib (PAC) versus best available therapy (BAT), including ruxolitinib (RUX), in patients (pts) with myelofibrosis (MF) l and platelet counts <100,000/µl. Blood. 2016;128:LBA-5.
CTI BioPharma. CTI BioPharma announces removal of full clinical hold on pacritinib. Press release. 2019. https://cbc.gcs-web.com/news-releases/news-release-details/cti-biopharma-announces-removal-full-clinical-hold-pacritinib. Accessed Sep 2019.
Geyer HL, Kosiorek H, Dueck AC, et al. Associations between gender, disease features and symptom burden in patients with myeloproliferative neoplasms: an analysis by the MPN QOL International Working Group. Haematologica. 2017;102:85–93.
Sunder RA. Financial toxicity: a growing burden for cancer patients. Bulletin American College Surgeons. 2019. https://bulletin.facs.org/2019/09/financial-toxicity-a-growing-burden-for-cancer-patients/. Accessed Dec 2019.
Parasuraman SV, Naim AB, Paranagama DC, et al. Financial burden of myeloproliferative neoplasms on patients: results from the MPN Landmark survey in the United States. Blood. 2015;126(23):5561.
Steensma DP, Komrokji RS, Stone RM, et al. Disparity in perceptions of disease characteristics, treatment effectiveness, and factors influencing treatment adherence between physicians and patients with myelodysplastic syndromes. Cancer. 2014;120:1670–6.
Xiao C, Polomano R, Bruner DW. Comparison between patient-reported and clinician-observed symptoms in oncology. Cancer Nurs. 2013;36:E1–E16.
MPN Research Foundation. Off-label drugs and the compendia. 2019. https://www.mpnresearchfoundation.org/Drug-Reimbursement. Accessed Sep 2019.
Drain PK, Parker RA, Robine M, Holmes KK, Bassett IV. Global migration of clinical research during the era of trial registration. PLoS One. 2018;13:e0192413 (Erratum in: PLoS One. 2018;13:e0199952).
ClinicalTrails.gov. Locations of registered studies. 2019. https://clinicaltrials.gov/ct2/resources/trends#LocationsOfRegisteredStudies. Accessed Sep 2019.
Okpechi IG, Swanepoel CR, Venter F. Access to medications and conducting clinical trials in LMICs. Nat Rev Nephrol. 2015;11:189–94.
NHS Cancer Patient Experience Survey 2011/12 National Report. 2019. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/212860/Cancer-Patient-Experience-Survey-National-Report-2011-12.pdf. Accessed Sep 2019.
Ford I, Norrie J. Pragmatic trials. N Engl J Med. 2016;375:454–63.
Hirsch IB, Martinez J, Dorsey ER, et al. Incorporating site-less clinical trials into drug development: a framework for action. Clin Ther. 2017;39:1064–76.
Scherber R, Dueck AC, Johansson P, et al. The myeloproliferative neoplasm symptom assessment form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118:401–8.
Acknowledgements
Funding
Development of the manuscript and payment of the Rapid Service and Open Access Fees were funded by Novartis Pharmaceuticals Corporation.
Medical Writing and Editorial Assistance
The authors received writing/editorial support in the preparation of this manuscript provided by Keisha Peters, MSc, of Excerpta Medica, funded by Novartis Pharmaceuticals Corporation.
Authorship
Both named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published. Both authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Disclosures
Cheryl Petruk and Jonathan Mathias have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12012594.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Petruk, C., Mathias, J. The Myeloproliferative Neoplasm Landscape: A Patient’s Eye View. Adv Ther 37, 2050–2070 (2020). https://doi.org/10.1007/s12325-020-01314-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01314-0