
Abstract
Adult T-cell leukemia–lymphoma (ATL), a rare and aggressive T-cell malignancy caused by human T-cell lymphotropic virus type 1 (HTLV-1), is associated with a poor prognosis. Evidence-based standard treatment options are lacking and outcomes are generally unsatisfactory, particularly for patients with relapsed or refractory disease. Continued research is contributing to changing treatment landscape as a number of existing and investigational agents are evaluated. We describe the epidemiology of HTLV-1 and ATL, discuss the biology behind the disease, review current treatment practices and guidelines, and provide an overview of emerging therapies in ATL, with a focus on those for relapsed or refractory disease.
Similar content being viewed by others


Introduction
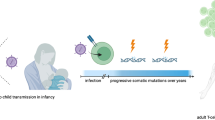
Adult T-cell leukemia–lymphoma (ATL) is a rare and aggressive peripheral T-cell neoplasm caused by human T-cell lymphotropic virus type 1 (HTLV-1) [1, 2]. ATL can present with diverse clinical features, but typically is associated with circulating leukemic cells, generalized lymph node swelling, hepatosplenomegaly, skin involvement, opportunistic infections, and hypercalcemia [3]. ATL generally has a poor prognosis, with shorter overall survival (OS) relative to other peripheral T-cell lymphomas (PTCLs) [4]. Factors contributing to poor outcomes include inherent chemoresistance and immunosuppression associated with ATL, particularly with aggressive forms [5]. Although progress has been made in understanding the biologic underpinnings of ATL, treatment outcomes generally remain unsatisfactory. Management of relapsed or primary refractory (R/R) ATL presents a particular challenge. The purpose of this review is to provide an overview of ATL (biology, epidemiology, diagnosis, and prognosis) and a brief review of current treatment guidelines, and to discuss emerging therapies, with a focus on those that may serve as viable treatment options for R/R ATL. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
HTLV-1 and ATL
ATL is caused by the HTLV-1 retrovirus [1, 2]. HTLV-1 causes transformation and clonal expansion of T cells, in some cases resulting in ATL [6]. Leukemogenesis of ATL is believed to be a multistep process involving a number of factors, including viral, epigenetic, and constitutional and acquired genetic factors and events [7, 8].
It is estimated that at least 5–10 million individuals worldwide are infected with HTLV-1 [9]. Infection with the HTLV-1 virus is endemic in several regions, including southwestern Japan, some parts of the Caribbean, South America, some areas of intertropical Africa (such as South Gabon) and of the middle East (such as the Mashad region in Iran), isolated clusters in Australo–Melanesia, and Romania (the only known region in Europe) [9]. In the USA and Europe, infection is usually seen in people from, or in sexual partners of people from, endemic regions [9,10,11].
Most individuals infected with HTLV-1 remain asymptomatic carriers; the lifetime risk of developing ATL among HTLV-1 carriers is estimated at 3–5% [12]. Currently there is no established method to prevent progression to ATL in HTLV-1 carriers, although several risk factors have been identified, such as host susceptibility factors, laboratory markers, and viral markers (reviewed by Iwanaga et al. [10]). In particular, proviral load appears to be a useful marker [13]. High proviral load is associated with increased risk of aggressive ATL [14].
The epidemiology of ATL reflects its association with HTLV-1. The geographic distribution of ATL corresponds with that of HTLV-1 carriers, with high incidence rates of ATL in HTLV-1 endemic regions [10]. For example, ATL accounts for approximately 25% of PTCLs in Asia (primarily Japan) compared with 2% in North America and 1% in Europe [4]. A recent study reported that ATL accounts for 5.5% of non-Hodgkin lymphoma (NHL) in Peru, 0.5% in Chile, and 1.1% in Central and South America overall [15]. The onset of ATL generally occurs following a latency period of approximately 30–50 years after initial HTLV-1 infection that occurs in most of the cases following breast feeding; as such, the disease primarily affects adults [11, 16,17,18]. The age of onset of ATL varies somewhat by geographic region; patients in the Caribbean or South America (40–50 years) and in the USA (approximately 50 years) tend to have a younger age at diagnosis than patients in Japan (where median age at diagnosis has increased from approximately 53 years in the 1980s to 66 years in 2006–2007) [10, 11, 19]. Regional differences have also been observed in the gender distribution of ATL. There is a clear male predominance in Japan but not in Jamaica [20] and one study from the USA showed a substantial female predominance [21].
Diagnosis and Prognosis
Diagnosis of ATL is determined by a combination of clinical presentation and morphologic/immunophenotypic features of the malignant cells, along with confirmation of HTLV-1 infection [17, 22]. Abnormal T cells characteristic of ATL have markedly polylobated nuclei with homogeneous and condensed chromatin, small or absent nucleoli, and agranular and basophilic cytoplasm [22]. The cells have a flower petal-like appearance and often express a CD3+ CD4+ CD5+ CD7− CD8− CD25+ immunophenotype [23]. ATL tumor cells are detected in peripheral blood or biopsy of affected organs [17]. At least 5% of circulating abnormal T lymphocytes are required for a diagnosis of ATL in patients without histologically proven tumor lesions [22].
ATL is classified into four subtypes based on the Shimoyama criteria; the acute and lymphoma subtypes are considered aggressive forms, while chronic and smoldering ATL have a more indolent course [24]. Generally, the aggressive disease types comprise the majority of ATL cases. For example, in Japan and Brazil, the acute type accounts for 55–60% of cases, lymphoma 20–25%, chronic 10–20%, and smoldering 5–10% [24,25,26]. However, data from the International Peripheral T-cell Lymphoma Project showed that 87% of aggressive ATL cases were the lymphoma type (13% were acute type) [27], suggesting that the lymphoma type might be more frequent than expected. In addition, some data indicate that the distribution may differ in other geographic regions [19, 28, 29].
The clinical features of ATL vary by disease type [24]. Acute-type ATL presents with a large number of circulating leukemia cells, generalized lymphadenopathy, hepatosplenomegaly, lytic bone lesions, visceral lesions, skin involvement, and systemic symptoms resulting from organ involvement, hypercalcemia, or opportunistic infection [3, 17]. The lymphoma type presents with lymphadenopathy in the absence of circulating leukemic cells in the peripheral blood. Patients may present with skin lesions, lung lesions, hepatomegaly, splenomegaly, and hypercalcemia, but these manifestations may be less frequent compared with the acute type [3, 17, 24]. Chronic-type ATL is associated with chronic peripheral lymphocytosis for several years and may occasionally be associated with skin and lung involvement, lymphadenopathy, and hepatosplenomegaly; no associated hypercalcemia or infiltration of the CNS, gastrointestinal tract, or bones are seen, and lactate dehydrogenase levels are normal or only slightly increased (less than twice the upper limit of normal) [3, 17, 24]. Chronic-type ATL may further be subdivided into favorable and unfavorable subtypes, based on clinical parameters [serum albumin, blood urea nitrogen (BUN), and lactate dehydrogenase (LDH) levels] [30]. The smoldering type characteristically shows skin or lung infiltration with no other visceral involvement, a normal lymphocyte count, and at least 5% abnormal lymphocytes in the peripheral blood [3, 17, 24]. Aggressive ATL types are associated with a particularly poor prognosis (median OS approximately 6–10 months); indolent types generally have a median OS of at least 2 years [19, 24, 25].
In addition to ATL disease type, several other prognostic factors have been identified. Recent evidence suggests there may be a higher frequency of poor prognostic factors among Caribbean patients compared with Japanese patients [31]. Factors that predict poor prognosis include poor performance status, elevated LDH level, at least four total involved lesions, hypercalcemia, age at least 40 years, thrombocytopenia, eosinophilia, bone marrow involvement, high interleukin-5 serum level, C–C chemokine receptor 4 (CCR4) expression, lung resistance-related protein, p53 mutation, and p16 deletion [22]. Risk models incorporating combinations of these factors have been developed that have shown utility in predicting outcomes [19, 27, 32, 33] and may provide further insight with regard to prognosis and/or treatment selection.
Current Treatment Landscape
Currently, there are no optimal standard treatment regimens for ATL. Most patients with ATL do not achieve a cure with current treatment options [3] and the efficacy of long-term therapy is limited [34]. Enrollment in clinical trials is commonly recommended, particularly for patients with R/R ATL, for whom existing treatment options are quite limited [3, 22, 34].
Consistent with the geographic distribution of HTLV-1 and ATL, most of the existing clinical trial data in ATL are based on studies conducted in Japan. Thus, the data must be extrapolated to predict responses in Western patients. Evidence of regional differences in ATL (e.g., distribution of ATL disease types [19, 24,25,26, 28] and frequency of poor risk factors [31], as mentioned earlier) suggests the possibility of differences in treatment outcomes for different populations. Further, differences in outcomes between clinical trials conducted in the USA [35, 36] and those conducted with similar regimens in Japan suggest that ATL may be more chemoresistant in the Western hemisphere. Ongoing clinical studies in Western patients will add much needed efficacy/safety data in this population, which may help to further refine treatment selection. In addition, studies of molecular events associated with HTLV-1 transformation to ATL in different populations will help to delineate these differences. However, it is important to note that most of the long-surviving patients in large series from Japan received allogeneic bone marrow transplantation [25].
Treatment strategies for ATL are based primarily on disease type, along with other prognostic factors and response to initial treatment [22, 34, 37]. In general, current treatment options for ATL include watchful waiting, zidovudine plus interferon-alfa (AZT/IFN), multi-agent chemotherapy, or allogeneic hematopoietic stem cell transplantation (allo-HSCT) [22, 34, 37]. Multi-agent chemotherapy may include the following combinations: CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone); CHOEP (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone); dose-adjusted EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin); hyper-CVAD (cyclophosphamide, vincristine, doxorubicin, and dexamethasone); or VCAP-AMP-VECP (vincristine, cyclophosphamide, doxorubicin, and prednisone; doxorubicin, ranimustine, and prednisone; and vindesine, etoposide, carboplatin, and prednisone). The selection of specific agents or combinations may vary by geographic region.
For first-line therapy, some international and US-based guidelines recommend the following approaches [22, 34, 37]. For chronic or smoldering ATL, observation may be appropriate for patients who are asymptomatic. Patients who are symptomatic (e.g., skin lesions, opportunistic infections) may be treated with skin-directed therapies or antiviral therapy (AZT/IFN), or may consider participation in a clinical trial. For unfavorable chronic or acute ATL, enrollment in a clinical trial is recommended; alternatively, treatment options include AZT/IFN or combination chemotherapy (e.g., CHOP, CHOEP, dose-adjusted EPOCH, hyper-CVAD, or VCAP-AMP-VECP). However, for chronic ATL, studies have shown that chemotherapies may worsen the prognosis when compared to watch and wait [38], whereas in the same situation, AZT/INF may induce long-term survival [29]. For the lymphoma subtype, enrollment in a clinical trial is again recommended; alternatively, patients may receive treatment with combination chemotherapy (as mentioned for acute ATL). In some cases, particularly in Japan, the mLSG15 regimen (VCAP-AMP-VECP) is recommended for aggressive ATL (i.e., acute or lymphoma subtype) [22, 39]. Similar regimens may be investigated in the USA.
For subsequent therapy, recommendations include the following options [22, 34]. For patients with acute or lymphoma types who achieve initial response to primary therapy, continuation of previous therapy or allogeneic hematopoietic stem cell transplantation (allo-HSCT) may be appropriate. Reports have shown that autologous transplantation is of little benefit for patients with ATL [40]. A recent retrospective analysis from Japan suggests that allo-HSCT at first remission may improve OS in some patients [25]. Patient age is a key factor in determining the appropriateness of allo-HSCT and the type of conditioning regimen used (i.e., myeloablative vs reduced intensity) [25]. The mean age of onset of ATL has increased in Japan [25] and is higher compared with other regions (e.g., the Caribbean) [10, 11]; thus, patterns of allo-HSCT use may vary over time and by location. In addition, the ability to find appropriate matched donors affects the use of allo-HSCT. The role of haplo-identical allo-HSCT in this context remains to be determined [41].
Patients with acute or lymphoma types who do not respond to primary therapy are a population with a significant unmet medical need; outcomes are dismal for these patients, and evidence-based treatments are lacking. Options for treatment of R/R ATL are very limited and might include a clinical trial, best supportive care, or an alternate therapy not previously used (such as AZT/IFN for acute or chemotherapy for lymphoma) [22, 34]. Registry studies from Japan indicate that allo-HSCT has been used in patients who were not in complete remission, although outcomes were poorer for patients who were not in complete remission compared with those who were [42, 43].
Emerging Treatments in R/R ATL
A number of new agents or combination therapies are currently being investigated for treatment of ATL. Below we review several emerging treatments that have ongoing or completed clinical trials that include patients with R/R disease, a population of significant unmet need. Agents are grouped by class and presented generally in chronological order. Information regarding clinical trials for emerging therapies in patients with R/R ATL is summarized in Table 1 [35, 44,45,46,47,48,49,50,51,52,53,54,55,56,57,58].
Antimetabolites
Cladribine
Cladribine is a purine nucleoside analogue resistant to degradation by adenosine deaminase [59, 60]. It is approved in the USA, European Union, and Japan for treatment of hairy cell leukemia [59, 60].
Phosphorylated derivatives of cladribine accumulate in lymphocytes with high deoxycytidine kinase activity, resulting in DNA strand breaks and cell death [44, 59, 60]. Cladribine is distinct among nucleoside analogues in that it is toxic in both rapidly proliferating cells and in resting cells [59, 60]. Cladribine was evaluated in a phase 2 trial in patients with R/R ATL (acute, lymphoma, or unfavorable chronic); however, it was terminated because of the low efficacy in futility analysis [44] (see Table 1).
Clofarabine
Clofarabine is a purine nucleoside metabolic inhibitor [61] that is structurally related to cladribine [62]. It is approved in the USA and European Union for treatment of pediatric patients 1–21 years old with R/R acute lymphoblastic leukemia after at least two prior regimens [61, 63].
Clofarabine inhibits DNA polymerases and ribonucleoside reductase; it also causes disruption of mitochondrial membrane integrity with release of cytochrome C and other apoptosis-inducing factors, leading to programmed cell death [61, 63]. A phase 1/2 study of the nucleoside analogue clofarabine in patients with R/R T-cell or natural killer (NK) cell lymphoma, including R/R ATL (NCT00416351), is ongoing.
Pralatrexate
Pralatrexate is a folate analogue metabolic inhibitor. It is currently approved in the USA for the treatment of patients with R/R PTCL [64].
Pralatrexate competitively inhibits dihydrofolate reductase and is a competitive inhibitor for polyglutamylation by the enzyme folylpolyglutamyl synthetase. This inhibition results in the depletion of thymidine and other biologic molecules [64].
The PROPEL study was a phase 2 study of pralatrexate in patients with R/R PTCL (however, the study population included only one patient with ATL) [65]. A phase 1/2a study evaluating pralatrexate plus the histone deacetylase (HDAC) inhibitor romidepsin in R/R lymphoid malignancies (including ATL) or multiple myeloma is currently recruiting (NCT01947140) (see Table 1).
Monoclonal Antibodies
Mogamulizumab
Mogamulizumab is a novel, defucosylated, humanized, monoclonal antibody targeting CCR4 [66]. CCR4 is one of the chemokine receptors involved in leukocyte migration and is selectively expressed in type 2 helper T cells (Th2) and regulatory T (Treg) cells [67]. CCR4 is often shown to be expressed in certain hematologic malignancies [67]. It is currently approved in Japan for the treatment of patients with treatment-naïve or R/R CCR4+ ATL as well as for R/R CCR4+ PTCL and cutaneous T-cell lymphoma (CTCL) [68]. Mogamulizumab in combination with dose-intensified chemotherapy has also demonstrated efficacy in newly diagnosed ATL [69].
Mogamulizumab demonstrates multiple potential mechanisms of action. It demonstrates potent antitumor activity and is mediated by highly enhanced antibody-dependent cellular cytotoxicity (ADCC) [70] because of its reduced fucose content [71]. Mogamulizumab also has been shown to deplete Treg cells, resulting in increased antitumor immune response [72, 73], thereby demonstrating activity as an immune-oncology agent. A recent translational study in patients with advanced or recurrent CCR4-negative solid cancers demonstrated that mogamulizumab depleted FoxP3+ CD4 Treg cells and that the effect was generally durable for more than 6 months after finishing eight infusions of mogamulizumab [74]. The depletion of Treg cells may explain why mogamulizumab use in the pre-transplantation setting has been associated with an increased risk of severe graft-versus-host disease (GVHD) and GVHD-related mortality [75, 76].
Completed clinical trials of mogamulizumab in ATL include a phase 1 trial in relapsed ATL/PTCL [46] and a phase 2 trial in relapsed ATL [47]. A phase 2 study in previously treated ATL vs investigator’s choice is currently ongoing [48] (see Table 1).
Daclizumab
Daclizumab is an anti-CD25 antibody [49]. It had been approved in the USA and European Union for prevention of acute allograft rejection, but manufacturing stopped in 2009. A new form (daclizumab high-yield process) is currently under review in the USA and European Union for treatment of relapsing multiple sclerosis.
Daclizumab acts by blocking CD25 (IL2R-α), thereby preventing the interaction of IL-2 with the high-affinity receptor and decreasing IL-2-mediated maintenance of the cytokine-dependent target cells [49]. A completed phase 2 trial evaluated daclizumab in patients with all subtypes of ATL (the majority of whom had received previous treatment) [49] (see Table 1).
Brentuximab Vedotin
Brentuximab vedotin is an antibody–drug conjugate (ADC). It is currently approved in the USA and European Union for treatment of patients with Hodgkin lymphoma (CD30+ HL, specifically, in the European Union) after failure of autologous stem cell transplant (ASCT) or after failure of at least two previous multi-agent chemotherapy regimens in patients who are not ASCT candidates, for patients with R/R systemic anaplastic large cell lymphoma (after failure of at least one previous multi-agent chemotherapy regimen in the USA), and in the USA for patients with primary cutaneous anaplastic large cell lymphoma or CD30+ mycosis fungoides (MF) who have received prior systemic therapy [77, 78].
Brentuximab vedotin is an anti-CD30 monoclonal antibody attached by a protease-cleavable linker to a cytotoxic agent, the microtubule-disrupting agent monomethyl auristatin E (MMAE). Following binding of the ADC to CD30-expressing cells and internalization of the ADC–CD30 complex, MMAE is released by proteolytic cleavage, resulting in targeted cell death via the microtubule-disrupting actions of MMAE [77,78,79].
Clinical trials include two ongoing trials in patients with R/R CD30+ lymphomas, including some ATL patients (a pilot study in patients with R/R disease [NCT01703949] and a phase 2 study in R/R CD30-low mature T-cell lymphomas [NCT01805037]) (see Table 1).
Alemtuzumab
Alemtuzumab is a CD52-directed cytolytic antibody approved in the USA for treatment of B-cell chronic lymphocytic leukemia [80].
Alemtuzumab binds to CD52, an abundant membrane glycoprotein expressed on the surface of B and T lymphocytes, monocytes, macrophages, and eosinophils [80, 81]. This binding results in complement-mediated lysis and ADCC through activation of NK cells and macrophages [80, 81]. A completed phase 2 study evaluated alemtuzumab in patients with chronic, acute, and lymphomatous ATL, the majority of whom had received prior treatment for ATL [50] (see Table 1).
Proteasome Inhibitor
Bortezomib
Bortezomib is a proteasome inhibitor approved in the USA and by the European Medicines Agency (EMA) for treatment of patients with multiple myeloma and patients with mantle cell lymphoma [82, 83].
Bortezomib reversibly inhibits activity of the 26S proteasome (a large protein complex that degrades ubiquitinated proteins), which prevents targeted proteolysis within the cell (including blockade of the degradation of IkBα, which prevents the activation of nuclear factor-kB [NF-kB] [51]), affecting multiple signaling cascades and ultimately leading to cell death [82, 83]. Bortezomib may also increase sensitivity of cancer cells to traditional anticancer agents. Clinical trials include a phase 2 trial with bortezomib monotherapy in R/R ATL (study terminated because of unpromising results) [51]; and a recently completed phase 1/2 trial of EPOCH with bortezomib and the integrase inhibitor raltegravir in previously treated or untreated ATL [35] (see Table 1).
Aurora A Kinase Inhibitor
Alisertib
Alisertib is an investigational Aurora A kinase (AAK) inhibitor. Alisertib causes G2/M arrest, abnormal mitotic spindle formation, the appearance of tetraploidy, and subsequent apoptosis [52]. A completed phase 2 clinical trial evaluated alisertib in patients with R/R PTCL (that included four patients with R/R ATL) [52]. A phase 1 trial of alisertib plus the HDAC inhibitor vorinostat in patients with R/R Hodgkin lymphoma, B-cell NHL, or PTCL (including ATL; NCT01567709) is ongoing (see Table 1).
Immunomodulatory Agents
Lenalidomide
Lenalidomide is a thalidomide analogue that is an immunomodulatory agent with antiangiogenic and antineoplastic properties [84]. It is currently approved by the US Food and Drug Administration (FDA) and the EMA for treatment of multiple myeloma (MM), transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes (MDS) associated with a deletion 5q abnormality with or without additional cytogenetic abnormalities [84, 85], and, in the USA, is also approved for treatment of relapsed or progressed mantle cell lymphoma (MCL) [84].
Lenalidomide inhibits proliferation and induces apoptosis of certain hematopoietic tumor cells, including multiple myeloma, mantle cell lymphoma, and del (5q) MDS, and causes a delay in tumor growth in some hematopoietic tumor models, including multiple myeloma. Immunomodulatory properties of lenalidomide include activation of T cells and NK cells, increased numbers of NKT cells, and inhibition of pro-inflammatory cytokines (e.g., TNF-α and IL-6) by monocytes [84, 85].
Completed clinical trials with lenalidomide in patients with R/R ATL include a multicenter, open-label, phase 1 study in patients with relapsed ATL (or PTCL) [55], a multicenter, open-label, phase 2 study in Japan [53], and a phase 2 study in North America [54] (see Table 1).
Therapeutic Vaccines
Therapeutic vaccines would offer a novel mechanism for treating ATL by means of stimulating an immune response against HTLV-1. Although still early in development, these agents may provide additional options for R/R ATL.
The Tax peptide-pulsed dendritic cell (Tax-DC) vaccine was designed to augment an HTLV-I Tax-specific cytotoxic T lymphocyte (CTL) response that has been implicated in anti-ATL effects [56]. It consists of autologous DCs pulsed with Tax peptides corresponding to the CTL epitopes [56]. Clinical trials include a pilot study of three patients with ATL who were previously treated and classified as intermediate to high risk [56].
THV02 is a therapeutic vaccine candidate for treatment of ATL. THV02 comprises two lentiviral vectors to be used in a prime/boost regimen [57]. THV02 encodes for a unique polypeptide derived from Tax, HBZ, p12I and p30II proteins, involved in HTLV-1 pathogenicity and known to be recognized by the immune system of HTLV-1 infected patients [86]. Preclinical results have demonstrated that THV02 can induce a cellular immune response in animal models [86]. THV02 was granted Orphan Drug Designation in 2015 by the EMA on the basis of preclinical immunogenicity results; clinical trials in ATL are planned [57].
Others
Agents with Clinical Trial Results in Patients with R/R ATL
Arsenic trioxide has been evaluated in combination with IFN in patients with R/R ATL (results summarized in Table 1) [45] and demonstrated efficacy in combination with AZT and IFN in patients with newly diagnosed chronic ATL [87, 88]. In lymphoma, the combination of IFN/AZT/arsenic increased the time to progression (Hermine et al., unpublished data). The anti-CD25 recombinant immunotoxin LMB-2 in combination with fludarabine and cyclophosphamide was evaluated in a phase 2 study in patients with CD25+ ATL, the majority of whom were previously treated (see Table 1) [58].
Agents with Completed (But Unpublished) or Ongoing Studies in R/R ATL
A phase 1 study of the proteasome inhibitor carfilzomib in patients with R/R T-cell lymphoma, including R/R ATL (NCT01336920) is ongoing (not recruiting). A phase 2 study of the immune toxin IMTOX-25 in patients with R/R CD25+ ATL (NCT01378871) has been completed; results have not yet been published. Two studies of the JAK inhibitor ruxolitinib are currently recruiting: a phase 2 study in patients with smoldering or chronic ATL [or previously treated lymphomatous or acute ATL with clinically indolent behavior indicated by lack of significant symptoms and treatment-free interval of greater than 6 months (NCT01712659)] and a phase 2 study in patients with R/R diffuse large B-cell or peripheral T-cell NHL, including ATL (NCT01431209). Studies of HDAC inhibitors include a phase 2 study of panobinostat in patients with R/R NHL (including recurrent ATL) that is currently ongoing (NCT01261247) and a phase 2 study of HBI-8000 in patients with R/R ATL that is currently recruiting (NCT02955589). A study of the PD-1 antibody pembrolizumab in R/R peripheral T-cell NHL (including ATL) is currently recruiting (NCT02535247). A phase 1 study of the dual PI3 K delta/gamma inhibitor RP6530 in patients with R/R peripheral (or cutaneous) T-cell lymphomas is currently recruiting (NCT02567656). See Table 1 for additional information on these studies.
Selected Agents with Studies in ATL and Related Conditions
Additional studies of HDAC inhibitors and checkpoint inhibitors in patients with ATL (not specifically R/R ATL) and other T-cell malignancies are also of interest. The HDAC inhibitors romidepsin and vorinostat have demonstrated efficacy in and are FDA approved for treatment of R/R cutaneous T-cell lymphoma [89,90,91,92]. Studies of HDAC inhibitors (in combination with AZT and/or IFN) in patients with ATL include a recently completed phase 1/2 trial that evaluated valproic acid in combination with AZT/IFN as maintenance therapy (NCT00854581) [93] and an ongoing study of belinostat in combination with AZT as consolidation therapy (NCT02737046) [94]. Clinical trials of checkpoint inhibitors include a phase 2 study of the PD-1 antibody nivolumab in patients with ATL (NCT02631746).
Conclusions
Patients with ATL are rarely cured with currently available cytotoxic drugs. Only those with chronic forms treated with antiviral therapy experienced long survival. In aggressive forms, only allo-HSCT after cytotoxic chemotherapy can cure some patients. The use of higher dose of chemotherapies and/or new cytotoxic agents did not significantly improve overall survival. At the present time, it is not yet clear whether increased response rates seen with combinations of monoclonal antibodies and chemotherapies could translate into an improvement of overall survival. Patients with R/R disease face a difficult prognosis and a limited number of treatment options. A number of different treatments have been and are being studied for use in R/R ATL, including several that are commercially available and approved for other indications. Emerging therapies with novel mechanisms of action and that target different pathways may further expand the number of available treatment options and improve outcomes for patients with R/R ATL. Therapies which aim to increase immune response may be of particular interest in this disease.
References
Poiesz BJ, Ruscetti FW, Reitz MS, Kalyanaraman VS, Gallo RC. Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cell leukaemia. Nature. 1981;294:268–71.
Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci USA. 1982;79:2031–5.
Tsukasaki K, Watanabe T, Tobinai K. Adult T-cell leukemia–lymphoma. In: Niederhuber JE, Armitage JO, Doroshow JH, et al., editors. Abeloff’s clinical oncology. 5th ed. Philadelphia: Elsevier Saunders; 2013. p. 2076–92.
Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124–30.
Marcais A, Suarez F, Sibon D, Frenzel L, Hermine O, Bazarbachi A. Therapeutic options for adult T-cell leukemia/lymphoma. Curr Oncol Rep. 2013;15:457–64.
Markham PD, Salahuddin SZ, Kalyanaraman VS, Popovic M, Sarin P, Gallo RC. Infection and transformation of fresh human umbilical cord blood cells by multiple sources of human T-cell leukemia–lymphoma virus (HTLV). Int J Cancer. 1983;31:413–20.
Tsukasaki K, Tobinai K. Human T-cell lymphotropic virus type I-associated adult T-cell leukemia–lymphoma: new directions in clinical research. Clin Cancer Res. 2014;20:5217–25.
Qayyum S, Choi JK. Adult T-cell leukemia/lymphoma. Arch Pathol Lab Med. 2014;138:282–6.
Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
Iwanaga M, Watanabe T, Yamaguchi K. Adult T-cell leukemia: a review of epidemiological evidence. Front Microbiol. 2012;3:322.
Proietti FA, Carneiro-Proietti ABF, Catalan-Soares BC, Murphy EL. Global epidemiology of HTLV-I infection and associated diseases. Oncogene. 2005;24:6058–68.
Ishitsuka K, Tamura K. Human T-cell leukaemia virus type I and adult T-cell leukaemia–lymphoma. Lancet Oncol. 2014;15:e517–26.
Manns A, Miley WJ, Wilks RJ, et al. Quantitative proviral DNA and antibody levels in the natural history of HTLV-I infection. J Infect Dis. 1999;180:1487–93.
Hodson A, Laydon DJ, Bain BJ, Fields PA, Taylor GP. Pre-morbid human T-lymphotropic virus type I proviral load, rather than percentage of abnormal lymphocytes, is associated with an increased risk of aggressive adult T-cell leukemia/lymphoma. Haematologica. 2013;98:385–8.
Laurini JA, Perry AM, Boilesen E, et al. Classification of non-Hodgkin lymphoma in Central and South America: a review of 1028 cases. Blood. 2012;120:4795–801.
Goncalves DU, Proietti FA, Ribas JG, et al. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin Microbiol Rev. 2010;23:577–89.
Bazarbachi A, Suarez F, Fields P, Hermine O. How I treat adult T-cell leukemia/lymphoma. Blood. 2011;118:1736–45.
Kaplan JE, Khabbaz RF. The epidemiology of human T-lymphotropic virus types I and II. Rev Med Virol. 1993;3:137–48.
Phillips AA, Shapira I, Willim RD, et al. A critical analysis of prognostic factors in North American patients with human T-cell lymphotropic virus type-1-associated adult T-cell leukemia/lymphoma: a multicenter clinicopathologic experience and new prognostic score. Cancer. 2010;116:3438–46.
Hisada M, Stuver SO, Okayama A, et al. Persistent paradox of natural history of human T lymphotropic virus type I: parallel analyses of Japanese and Jamaican carriers. J Infect Dis. 2004;190:1605–9.
Levine PH, Dosik H, Joseph EM, et al. A study of adult T-cell leukemia/lymphoma incidence in central Brooklyn. Int J Cancer. 1999;80:662–6.
Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia–lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27:453–9.
Dasanu CA. Newer developments in adult T-cell leukemia/lymphoma therapeutics. Expert Opin Pharmacother. 2011;12:1709–17.
Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–1987). Br J Haematol. 1991;79:428–37.
Katsuya H, Ishitsuka K, Utsunomiya A, et al. Treatment and survival among 1594 patients with ATL diagnosed in the 2000s: a report from the ATL-PI project performed in Japan. Blood. 2015;126:2570–7.
Pombo De Oliveira MS, Loureiro P, Bittencourt A, et al. Geographic diversity of adult t-cell leukemia/lymphoma in Brazil. Int J Cancer. 1999;83:291–8.
Suzumiya J, Ohshima K, Tamura K, et al. The international prognostic index predicts outcome in aggressive adult T-cell leukemia/lymphoma: analysis of 126 patients from the international peripheral T-cell lymphoma project. Ann Oncol. 2009;20:715–21.
Hanchard B. Adult T-cell leukemia/lymphoma in Jamaica: 1986–1995. J Acquir Immun Defic Syndr Hum Retrovirol. 1996;13(suppl 1):S20–5.
Bazarbachi A, Plumelle Y, Carlos RJ, et al. Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol. 2010;28:4177–83.
Tsukasaki K, Tobinai K. HTLV-1-associated T-cell diseases. In: Francine F, editor. T-cell lymphomas. New York: Humana; 2013. p. 113–35.
Zell MI, Assal A, Konda B, et al. Analysis of large cohort shows that Caribbean adult T cell leukemia/lymphoma is a chemotherapy refractory disease with very poor prognosis that behaves distinctly from Japanese subtypes. Blood. 2014;124:1685.
A predictive model for aggressive non-Hodgkin’s lymphoma. The international non-Hodgkin’s lymphoma prognostic factors project. N Engl J Med. 1993;329:987–94.
Fukushima T, Nomura S, Shimoyama M, et al. Japan Clinical Oncology Group (JCOG) prognostic index and characterization of long-term survivors of aggressive adult T-cell leukaemia-lymphoma (JCOG0902A). Br J Haematol. 2014;166:739–48.
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): non-Hodgkin’s lymphomas. Version 2.2015, 2015.
Ratner L, Rauch D, Abel H, et al. Dose-adjusted EPOCH chemotherapy with bortezomib and raltegravir for human T-cell leukemia virus-associated adult T-cell leukemia lymphoma. Blood Cancer J. 2016;6:e408.
Ratner L, Harrington W, Feng X, et al. Human T cell leukemia virus reactivation with progression of adult T-cell leukemia–lymphoma. PLoS One. 2009;4:e4420.
British Committee for Standards in Haematology. Guidelines for the management of mature T-cell and NK-cell neoplasms (excluding cutaneous T-cell lymphoma). http://www.guideline.gov/content.aspx?id=47071. Accessed October 30, 2017.
Takasaki Y, Iwanaga M, Imaizumi Y, et al. Long-term study of indolent adult T-cell leukemia–lymphoma. Blood. 2010;115:4337–43.
Tsukasaki K, Utsunomiya A, Fukuda H, et al. VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia–lymphoma: Japan Clinical Oncology Group Study JCOG9801. J Clin Oncol. 2007;25:5458–64.
Tsukasaki K, Maeda T, Arimura K, et al. Poor outcome of autologous stem cell transplantation for adult T cell leukemia/lymphoma: a case report and review of the literature. Bone Marrow Transplant. 1999;23:87–9.
Phillips EH, Hodson A, Hermine O, Bazarbachi A, Cwynarski K. Striving to cure adult T-cell leukaemia/lymphoma: a role for allogeneic stem cell transplant? Bone Marrow Transplant. 2016;51:1549–55.
Hishizawa M, Kanda J, Utsunomiya A, et al. Transplantation of allogeneic hematopoietic stem cells for adult T-cell leukemia: a nationwide retrospective study. Blood. 2010;116:1369–76.
Ishida T, Hishizawa M, Kato K, et al. Allogeneic hematopoietic stem cell transplantation for adult T-cell leukemia–lymphoma with special emphasis on preconditioning regimen: a nationwide retrospective study. Blood. 2012;120:1734–41.
Tobinai K, Uike N, Saburi Y, et al. Phase II study of cladribine (2-chlorodeoxyadenosine) in relapsed or refractory adult T-cell leukemia–lymphoma. Int J Hematol. 2003;77:512–7.
Hermine O, Dombret H, Poupon J, et al. Phase II trial of arsenic trioxide and alpha interferon in patients with relapsed/refractory adult T-cell leukemia/lymphoma. Hematol J. 2004;5:130–4.
Yamamoto K, Utsunomiya A, Tobinai K, et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia–lymphoma and peripheral T-cell lymphoma. J Clin Oncol. 2010;28:1591–8.
Ishida T, Joh T, Uike N, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia–lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30:837–42.
Phillips A, Fields P, Hermine O, et al. A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab versus investigator’s choice in the treatment of patients with relapsed/refractory adult T-cell leukemia–lymphoma: overall response rate, progression-free survival, and overall survival. Blood. 2016;128:4159 (abstract).
Berkowitz JL, Janik JE, Stewart DM, et al. Safety, efficacy, and pharmacokinetics/pharmacodynamics of daclizumab (anti-CD25) in patients with adult T-cell leukemia/lymphoma. Clin Immunol. 2014;155:176–87.
Sharma K, Janik J, O’Mahony D, et al. Phase II study of alemtuzumab (CAMPATH-1®) in patients with HTLV-1-associated adult T-cell leukemia/lymphoma. Clin Cancer Res. 2017;23:35–42.
Ishitsuka K, Utsunomiya A, Katsuya H, et al. A phase II study of bortezomib in patients with relapsed or refractory aggressive adult T-cell leukemia/lymphoma. Cancer Sci. 2015;106:1219–23.
Barr PM, Li H, Spier C, et al. Phase II intergroup trial of alisertib in relapsed and refractory peripheral T-cell lymphoma and transformed mycosis fungoides: SWOG 1108. J Clin Oncol. 2015;33:2399–404.
Ishida T, Fujiwara H, Nosaka K, et al. Multicenter phase II study of lenalidomide in relapsed or recurrent adult T-cell leukemia/lymphoma: ATLL-002. J Clin Oncol. 2016;34:1219–23.
Phillips AA, Giddings J, Lee SM, Horwitz SM. Lenalidomide in patients with relapsed or refractory HTLV-1 related adult T cell leukemia/lymphoma (ATLL). Int J Blood Res Disorders. 2015;2:1–3.
Ogura M, Imaizumi Y, Uike N, et al. Lenalidomide in relapsed adult T-cell leukaemia-lymphoma or peripheral T-cell lymphoma (ATLL-001): a phase 1, multicentre, dose-escalation study. Lancet Haematol. 2016;3:e107–18.
Suehiro Y, Hasegawa A, Iino T, et al. Clinical outcomes of a novel therapeutic vaccine with Tax peptide-pulsed dendritic cells for adult T cell leukaemia/lymphoma in a pilot study. Br J Haematol. 2015;169:356–67.
Theravectys. Theravectys obtains orphan drug designation from the European Medicines Agency for its lentiviral vector-based therapeutic vaccine against adult T-cell leukemia and lymphoma [press release]. http://www.theravectys.com/wp-content/uploads/PDF/14_Theravectys_obtains_Orphan_Drug_Designation_from_EMA.pdf. Accessed October 30, 2017.
Kreitman RJ, Stetler-Stevenson M, Jaffe ES, et al. Complete remissions of adult T-cell leukemia with anti-CD25 recombinant immunotoxin LMB-2 and chemotherapy to block immunogenicity. Clin Cancer Res. 2016;22:310–8.
Leustatin® (cladribine) injection [prescribing information]. Raritan, NJ: Centocor Ortho Biotech Products, L.P.; July, 2012.
Litak® 2 mg/ml solution for injection [summary of product characteristics]. Weil/Rhein, Germany: Lipomed GmbH; August, 2013.
Clolar® (clofarabine) injection, for intravenous use [prescribing information]. Cambridge, MA: Genzyme Corporation; October, 2016.
Corey SJ. New agents in the treatment of childhood leukemias and myelodysplastic syndromes. Curr Oncol Rep. 2005;7:399–405.
Evoltra® 1 mg/ml concentrate for solution for infusion [summary of product characteristics]. The Netherlands: Genzyme Europe B.V. October, 2016.
Folotyn® (pralatrexate injection) [prescribing information]. Westminster, CO: Allos Therapeutics, Inc.; May, 2012.
O’Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29:1182–9.
Subramaniam JM, Whiteside G, McKeage K, Croxtall JC. Mogamulizumab: first global approval. Drugs. 2012;72:1293–8.
Ishida T, Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. 2006;97:1139–46.
Approval for additional indication for PTCL and CTCL of mogamulizumab. http://www.kyowa-kirin.com/news_releases/2014/e20140317_01.html. Accessed October 30, 2017.
Ishida T, Jo T, Takemoto S, et al. Dose-intensified chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: a randomized phase II study. Br J Haematol. 2015;169:672–82.
Ishii T, Ishida T, Utsunomiya A, et al. Defucosylated humanized anti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell leukemia/lymphoma. Clin Cancer Res. 2010;16:1520–31.
Shinkawa T, Nakamura K, Yamane N, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–73.
Sugiyama D, Nishikawa H, Maeda Y, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA. 2013;110:17945–50.
Ni X, Jorgensen JL, Goswami M, et al. Reduction of regulatory T cells by mogamulizumab, a defucosylated anti-CC chemokine receptor 4 antibody, in patients with aggressive/refractory mycosis fungoides and Sézary syndrome. Clin Cancer Res. 2015;21:274–85.
Kurose K, Ohue Y, Wada H, et al. Phase Ia study of FoxP3 + CD4 treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res. 2015;21:4327–36.
Fuji S, Inoue Y, Utsunomiya A, et al. Pretransplantation anti-CCR4 antibody mogamulizumab against adult T-cell leukemia/lymphoma is associated with significantly increased risks of severe and corticosteroid-refractory graft-versus-host disease, nonrelapse mortality, and overall mortality. J Clin Oncol. 2016;34:3426–33.
Sugio T, Kato K, Aoki T, et al. Mogamulizumab treatment prior to allogeneic hematopoietic stem cell transplantation induces severe acute graft-versus-host disease. Biol Blood Marrow Transplant. 2016;22:1608–14.
Adcetris® (brentuximab vedotin) [prescribing information]. Bothell, WA: Seattle Genetics, Inc.; November, 2017.
Adcetris® 50 mg powder for concentrate for solution for infusion [summary of product characteristics]. Taastrup, Denmark: Takeda Pharma A/S; May, 2015.
Fanale MA, Horwitz SM, Forero-Torres A, et al. Brentuximab vedotin in the front-line treatment of patients with CD30+ peripheral T-cell lymphomas: results of a phase I study. J Clin Oncol. 2014;32:3137–43.
Campath® (alemtuzumab) [prescribing information]. Cambridge, MA: Genzyme Corporation; September, 2014.
Ravandi F, O’Brien S. Alemtuzumab. Expert Rev Anticancer Ther. 2005;5:39–51.
Velcade® (bortezomib) for injection [prescribing information]. Cambridge, MA: Millennium Pharmaceuticals, Inc.; June, 2008.
Velcade® 1 mg powder for solution for injection [summary of product characteristics]. Beerse, Belgium: Janssen-Cilag International NV; March, 2015.
Revlimid® (lenalidomide) [prescribing information]. Summit, NJ: Celgene Corporation; 2007.
Revlimid® 2.5 mg hard capsules [summary of product characteristics]. Uxbridge, United Kingdom: Celgene Europe Limited; March, 2015.
Revaud D, Bejanariu A, Loussaief L, et al. Development of an anti-HTLV-1 vaccine for the treatment of adult T-cell leukemia/lymphoma. Presented at: 57th Annual Meeting and Exposition of the American Society of Hematology, December 5–8, 2015, Orlando, FL.
Kchour G, Tarhini M, Kooshyar MM, et al. Phase 2 study of the efficacy and safety of the combination of arsenic trioxide, interferon alpha, and zidovudine in newly diagnosed chronic adult T-cell leukemia/lymphoma (ATL). Blood. 2009;113:6528–32.
Kchour G, Rezaee R, Farid R, et al. The combination of arsenic, interferon-alpha, and zidovudine restores an “immunocompetent-like” cytokine expression profile in patients with adult T-cell leukemia lymphoma. Retrovirology. 2013;10:91.
Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–91.
Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–15.
Istodax® (romidepsin) for injection, for intravenous use [prescribing information]. Summit, NJ: Celgene Corporation; July, 2016.
Zolinza® (vorinostat) capsules [prescribing information]. Whitehouse Station, NJ: Merck & Co., Inc.; December, 2015.
Ramos JC, Toomey N, Diaz L, Ruiz P, Barber G, Harrington W Jr. Targeting HTLV-I latency in adult T-cell leukemia/lymphoma [abstract]. Retrovirology. 2011;8(suppl 1):16 (Abstract A48).
Toomey N, Ramos JC. The combination of belinostat with zidovudine for treatment of HTLV-I related adult T-cell leukemia–lymphoma. Presented at: 57th American Society of Hematology Annual Meeting and Exposition, December 5–8, 2015, Orlando, FL.
Acknowledgements
Funding
Funding to support the preparation of this manuscript, article processing charges, and open access fee was provided by Kyowa Kirin International (Bedminster, NJ, USA), a subsidiary of Kyowa Hakko Kirin Co. Ltd.
Medical Writing, Editorial, and Other Assistance
The authors thank Sherri D. Jones of MedVal Scientific Information Services, LLC for medical writing and editorial assistance, for which funding was provided to MedVal by Kyowa Kirin International, a subsidiary of Kyowa Hakko Kirin Co. Ltd. This manuscript was prepared according to the International Society for Medical Publication Professionals’ “Good Publication Practice for Communicating Company-Sponsored Medical Research: The GPP3 Guidelines.”
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published. All authors contributed equally and each were involved in data analysis/interpretation and in drafting or critically revising the manuscript.
Disclosures
Olivier Hermine has nothing to declare. Juan Carlos Ramos has nothing to declare. Kensei Tobinai received research funding from Kyowa Hakko Kirin, Celgene, Eisai, Mundipharma, Takeda; and has received honoraria from Eisai, Takeda, Mundipharma, HUYA Bioscience International, Kyowa Hakko Kirin, Celgene.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/212DF06002DB3474.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hermine, O., Ramos, J.C. & Tobinai, K. A Review of New Findings in Adult T-cell Leukemia–Lymphoma: A Focus on Current and Emerging Treatment Strategies. Adv Ther 35, 135–152 (2018). https://doi.org/10.1007/s12325-018-0658-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-018-0658-4