
Abstract
The novel beta coronavirus (SARS-CoV-2, designated as COVID-19) that is responsible for severe acute respiratory syndrome has devastated the global economy and health care system. Since COVID-19 changed the definition of “normal” in ordinary life around the world, the development of effective therapeutics and preventive measures is desperately needed to fight SARS-CoV-2 infection and restore normalcy. A clear understanding of COVID-19 pathogenesis is crucial in providing the scientific rationale necessary to develop anti-COVID19 drugs and vaccines. According to the most recently published literature, COVID-19 pathogenesis was postulated to occur in three sequential phases: pulmonary, proinflammatory, and prothrombic. Herein, virus-host interactions, potential pathogenic mechanisms, and clinical manifestations are described for each phase. Additionally, based on this pathogenesis model, various therapeutic strategies involving current clinical trials are presented with an explanation of their modes of action and example drugs. This review is a thorough, updated summary of COVID-19 pathogenesis and the therapeutic options available for this disease.
Similar content being viewed by others


Introduction
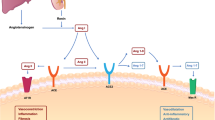
Coronavirus disease 2019 (COVID-19) has emerged as a full-fledged global pandemic after starting as an isolated infection of an unknown nature in Wuhan, China (December 2019). A new beta coronavirus strain called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was determined to be the causative agent for this unprecedented infectious disease. The COVID-19 pandemic has devastated the lives of millions of people around the world. At the same time, its negative impacts on the global economy and health care system have been unfathomable and are still ongoing. To develop effective therapeutics and preventive measures against COVID-19, an accurate and precise understanding of its pathogenesis at the molecular level is in absolute need. Based on the most recently published literature, the overall COVID-19 pathogenesis process can be summarized as three clinically distinct and potentially overlapping phases. As shown in Fig. 1, its pathological progression can be classified in chronological order as pulmonary, proinflammatory, and prothrombic phases (Domingo et al. 2020).
In the pulmonary phase, SARS-CoV-2 infects host target cells and causes interstitial pneumonia and acute respiratory distress syndrome (ARDS) through angiotensin-converting enzyme 2 (ACE2) deficiency and a subsequent renin-angiotensin system (RAS) imbalance. In the pro-inflammatory phase, infected target cells and lymphocytes further respond to SARS-CoV-2 infection by overproducing proinflammatory cytokines, which results in acute lung injury (ALI) and cytokine storm via systemic inflammation. In the final prothrombic phase, widespread platelet aggregation and thrombosis give rise to coagulopathy and multi-organ failure (MOF) at the end stage of COVID19 patients. Based on this three-step pathogenesis model, various phase-specific therapeutic strategies were classified with an emphasis on their mechanisms of action with corresponding examples.
Pulmonary phase
ACE2 deficiency
SARS-CoV-2 infects alveolar epithelial cells in both the upper and lower respiratory tracts. In general, alveolar epithelial cells are composed of type 1 and 2 pneumocytes. Type 1 pneumocytes, which describes 95% of all alveolar epithelial cells, are responsible for alveolar gas exchange. On the other hand, type 2 pneumocytes produce alveolar surfactants that reduce pulmonary surface tension. They also serve as stem cell populations that can differentiate into type 1 pneumocytes.
ACE2 is a type I integral membrane protein with an enzymatic activity of mono carboxypeptidase. Its amino acid sequence exhibits 46% homology with an angiotensin-converting enzyme (ACE) (Donoghue et al. 2000; Jiang et al. 2014). ACE2 cleaves the carboxyl-terminal amino acid phenylalanine from angiotensin II to produce the vasodilator angiotensin 1–7 (Donoghue et al. 2000). Since SARS-CoV-2 uses angiotensin-converting enzyme 2 (ACE2) as the main receptor for host entry (Dhochak et al. 2020), type 2 pneumocytes are regarded as the primary target of SARS-CoV-2 infection due to their overexpression of ACE 2 (Barkauskas et al. 2013; Rico-Mesa et al. 2020). More specifically, the interaction between the receptor-binding domain of the viral spike protein with ACE2 initiates the host entry process (Fig. 2a). Once engaged with ACE2, the ACE2-bound viral spike protein undergoes proteolytic cleavage, which is catalyzed by a host membrane-anchored protein called the receptor transmembrane protease serine 2 (TMPRSS2) (Glowacka et al. 2011). This host protease-assisted digestion induces a conformational change in the spike protein that is necessary for host and virus membrane fusion. After this spike-mediated fusion process, the internalized virus particle releases its RNA genome and begins replication. The co-internalized ACE2 receptor either undergoes intracellular degradation or recycles back to the plasma membrane. As a result, the continuous infection of host target cells by SARS-CoV-2 facilitates ACE2 downregulation and ultimately leads to chronic ACE2 deficiency (Fig. 2a) (Vaduganathan et al. 2020).
RAS imbalance
RAS is one of the most well-known physiological systems responsible for controlling cardiovascular dynamics through blood pressure modulation. To precisely modify this system, angiotensinogen is first converted into angiotensin I by an enzyme called renin, which is produced in the kidneys (Fig. 2b). This newly generated angiotensin I is further processed into angiotensin II by an extracellular angiotensin-converting enzyme (ACE). ACE is metalloproteinase distinct from the previously described SARS-CoV-2 receptor, ACE2. Angiotensin II binds to the G-protein coupled receptor (GPCR) angiotensin II type 1 receptor (AT1R) to initiate its various physiological functions as shown in Fig. 2b. Generally, activation of AT1R by angiotensin II causes several physiologically important events to occur including vasoconstriction, inflammation, fibrosis, thrombosis, and reactive oxygen species (ROS) production. On the other hand, angiotensin II is further degraded into angiotensin 1–7 by the enzymatic action of ACE2 (Fig. 2b). Angiotensin 1–7 binds to another GPCR called MasR and induces physiological events that are essentially opposite to those induced by AT1R activation. They include vasodilation, anti-inflammation, anti-fibrosis, anti-thrombosis, and ROS neutralization. In essence, ACE2 plays a key role as a negative regulator in the overall RAS pathway. Therefore, ACE2 exerts protective functions in various RAS-based pathogenesis models involving acute and chronic inflammation (Banu et al. 2020). ACE2 also limits macrophages from expressing several proinflammatory cytokines. They include tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), which are critical inflammatory components of COVID-19 pathogenesis, and they will be discussed later (Patel et al. 2016).
SARS-CoV-2-induced ACE2 downregulation and its subsequent deficiency continuously block the conversion of angiotensin II into angiotensin 1–7. As a result, excessive angiotensin II is constantly available for AT1R hyperactivation. This model is further supported by a loss of pulmonary ACE2 function associated with acute lung injury (ALI) and heart failure in other RAS-based disease models (Kuba et al. 2005, 2006; Imai et al. 2005, 2008). Moreover, angiotensin 1–7 also exhibits anti-inflammatory activities in the vascular system by decreasing levels of pro-inflammatory proteins such as vascular cell adhesion molecule 1 (VCAM-1), IL-6, and chemokine (C-C motif) ligand 2 (CCL2) (Zhang et al. 2015). Thus, SARS-CoV-2-induced ACE2 deficiency can also antagonize the anti-inflammatory properties of angiotensin 1–7. In support of this hypothesis, SARS-CoV-2-infected patients showed a significant increase in angiotensin II plasma levels. These enhanced angiotensin II plasma levels were inversely correlated with viral load (Liu et al. 2020c). As a result, ACE2 deficiency and a consequent RAS imbalance are considered one of the most clinically relevant molecular mechanisms responsible for the overall progression of COVID-19 pathogenesis (Henry et al. 2020).
Since pre-existing chronic inflammatory conditions such as hypertension, diabetes, obesity, cancer, cardiovascular disease, and autoimmune disease also activate the RAS pathway, COVID-19 patients exhibit a significant association between ACE2 deficiency and clinical severity of these comorbidities (Verdecchia et al. 2020; Tseng et al. 2020). The reduced expression of ACE2 with age also has clinical implications for the poor prognosis of elderly COVID-19 patients. Since the ACE2 gene is located on the X chromosome, it has been hypothesized that a higher male mortality rate in COVID-19 patients may be linked to lower expression levels of the ACE2 gene in male patients. Therefore, restoring the SARS-CoV-2-induced RAS imbalance has been suggested as an ideal clinical approach to slow the early progression of COVID-19 pathogenesis.
Aggravation of ACE2 deficiency
In addition to the primary ACE2 deficiency elicited by SARS-CoV-2 infection, two other mechanistically plausible ways to enhance the SARS-CoV-2-induced RAS imbalance exist. ADAM metallopeptidase domain 17 (ADAM-17) is a TNF-α-converting enzyme. It activates TNF-α by cleaving its extracellular domain, which converts it from its inactive membrane-bound form into its active, soluble form (Fig. 2c). Since AT1R stimulation by angiotensin II indirectly induces ADAM-17 activation, ADAM-17 overactivation may be an aggravating factor in COVID-19 pathogenesis (Patel et al. 2014; Aleksova et al. 2020). ADAM-17 overactivation by angiotensin II caused by virus-induced ACE2 deficiency leads to TNF-α overproduction. Since TNF-α is a primary cytokine implicated in the pathogenesis of many chronic inflammatory diseases, it may further exacerbate inflammatory conditions of COVID-19 patients by enhancing macrophage mobilization and vascular membrane permeability (Haga et al. 2008; Glowacka et al. 2010). Furthermore, ADAM-17 can also inactivate ACE2 by digesting its extracellular domain (Aleksova et al. 2020) (Fig. 2c). Following this digestion, truncated ACE2 is released into the extracellular space, further promoting ACE2 deficiency and increasing inflammation in COVID-19 patients (Black et al. 1997; Moss et al. 1997; Gheblawi et al. 2020).
ADAM-17 also seems to play a direct role in coronavirus entry; its enzymatic activity is stimulated when SARS-CoV-2 binds to the ACE2 receptor. This enhanced protolytic activity of ADAM-17 seems to facilitate viral entry (Haga et al. 2008). In addition to TNF-α activation, ADAM-17 also liberates several other membrane-bound cytokine precursors including IL-4 and interferon γ (IFN-γ) (Wang et al. 2020c). This could exert more detrimental effects on the proinflammatory reactions that are already upregulated by SARS-CoV-2 infection.
Des-arginine(9)-bradykinin (des-Arg9-BK) overproduction is another potential aggravating factor in COVID-19 pathogenesis. Des-Arg9-BK is a potent kinin-kallikrein system metabolite that increases vascular permeability. Thus, it promotes angioedema by acting on the type B1 BK receptor. This type of B1 BK receptor is, in turn, upregulated by other inflammatory cytokines (Vickers et al. 2002). Since ACE2 is the main enzyme responsible for des-Arg9-BK’s metabolic degradation (Fig. 2c) (Turner et al. 2004), SARS-CoV-2-induced ACE2 deficiency can cause increased des-Arg9-BK accumulation. This can further result in BK-B1 receptor-mediated signaling pathway overactivation (Fig. 2c). Pathologically, this could lead to increased angioedema, blood coagulation, and complementary pathway stimulation, all of which can further aggravate the proinflammation conditions induced by SARS-CoV-2 infection (Mahmudpour et al. 2020).
Proinflammatory phase
Activation of innate immunity
Pathogen-associated molecular patterns (PAMPs) recognition by host pattern recognition receptors (PRRs) is the first step in innate immune system activation against viral infection. These innate immune receptors include retinoic acid-inducible gene I (RIG-I)-like receptors, nucleotide-binding oligomerization domain (NOD)-like receptors, melanoma differentiation-associated gene 5 (MDA5), and Toll-like receptors (TLRs) (Fig. 3a). For RNA virus recognition, they directly recognize RNA virus-specific motifs such as 5′-triphosphate and double-stranded RNAs as PAMPs (Kowalinski et al. 2011; Nikonov et al. 2013). This virus-derived PAMP recognition by innate immune receptors activates a series of signaling cascades that ultimately lead to the activation of transcription factors such as nuclear factor-kappa B (NF-kB) and interferon regulatory factors (IRFs) (Fig. 3a). Their transcriptional activation of immune-related target genes leads to the production of type I and III IFNs and various proinflammatory cytokines and chemokines. Once secreted from cells, they exert diverse immunological effects on uninfected neighboring cells by binding to their corresponding cell surface receptors.
Endogenous self-antigens called damage-associated molecular patterns (DAMPs) also play an important role in innate immune system activation. Pyroptosis is a highly inflammatory form of programmed cell death that is commonly induced by cytopathic viruses (Fink and Cookson 2005). This unique process generates proinflammatory molecules with DAMPs. In general, DAMPs are recognized by immune receptors as endogenous danger signals induced by pathogenic microbes. This recognition also leads to the activation of NF-κB and IRF signaling pathways, which further drives the innate immune system to induce unscheduled cell death following microbial invasion (Fig. 3a) (Tang et al. 2012; Liu et al. 2017). In particular, NLR family pyrin domain containing 3 (NLRP3) protein (a NOD-like receptor) forms the inflammasome complex upon DAMP recognition. This DAMP recognition activates enzymatically inactive pro-caspase 1 into its enzymatically active form. Activated caspase 1 converts IL-1β and IL-18 inflammatory cytokine precursors into active ones. In vitro studies have demonstrated that a SARS-CoV-1 infection can induce DAMPs-initiated intracellular stress pathways, resulting in NLRP3-dependent inflammasome activation and macrophage pyroptosis (Shi et al. 2019; Chen et al. 2019; Soy et al. 2020).
It has been hypothesized that strong recognition of SARS-CoV-2 RNA elements by various innate receptors and subsequent target gene transcriptional activation causes IFN, cytokine, and chemokine overproduction (Fig. 3a). In this regard, a diverse array of cytokines are induced by SARS-CoV-2 infection (Coperchini et al. 2020; Gao et al. 2020; Mahmudpour et al. 2020; Picchianti Diamanti et al. 2020; Ragab et al. 2020; Soy et al. 2020; Wang et al. 2020a; Ye et al. 2020). They include IL-1β, -2, -4, -6, -7, -8, -10, -12, -17, -18, -21, -22, and − 33; tumor growth factor-beta (TGF-β); IFN-α, -β, and -γ; granulocyte colony-stimulating factor (G-CSF); granulocyte-macrophage colony-stimulating factor (GM-CSF); macrophage inflammatory protein 1 alpha (MIP-1α) and beta (MIP-1β); matrix metalloproteinase-1 (MMP-1) and MMP-3; TNF-α; and C-reactive protein (CRP) (Scala and Pacelli 2020). A number of chemokines are also induced by SARS-CoV-2 infection (Chen and Subbarao 2007; Channappanavar and Perlman 2017; Coperchini et al. 2020; Li et al. 2020). They include chemokine (C–C motif) ligand 2 (CCL2; also known as monocyte chemoattractant protein 1, MCP1), CCL3 and 5, C-X-C motif chemokine ligand 8 (CXCL-8; also known as interferon gamma-induced protein 10, IP-10), and CXCL-9 and 10 (Chen and Subbarao 2007; Scala and Pacelli 2020). Alveolar macrophages, lung epithelial cells, and dendritic cells have been suggested as the major cell types that produce these proinflammatory cytokines and chemokines. However, an unusual lack of IFN response to SARS-CoV-2 infection was also noticed in SARS-CoV-2-infected cells in vitro (Chen and Subbarao 2007). In line with this, IFN can only be detected after SARS-CoV reached a high titer in coronavirus patients. Based on these observations, it has been postulated that SARS-CoV may delay or evade the innate immune response through early antagonism of the IFN response (Yoshikawa et al. 2010; Channappanavar et al. 2016; Channappanavar and Perlman 2017). Several studies are underway to elucidate the potential molecular mechanisms SARS-CoV-2 employs to suppress IFN activation.
Lymphopenia
Lymphopenia is defined as a dramatic reduction in overall immune cell numbers circulating in the blood. COVID-19 patients exhibit general lymphopenia in dendritic cells, macrophages, and T-cells (Sallenave and Guillot 2020). SARS-CoV-2 infection also reduces circulating NK cell numbers and causes an exhausted phenotype in NK cells (Market et al. 2020). The increased expression of NKG2A, programmed cell death protein 1 (PD-1), and T cell membrane protein 3 (TIM-3) further indicate viral-induced T cell and NK cell exhaustion after SARS-CoV-2 infection (Paces et al. 2020). The virus’s ability to kill lymphocytes could contribute to the observed lymphopenia in coronavirus patients (Gu et al. 2005). Although lymphopenia, leukopenia, and thrombocytopenia are laboratory parameters that are consistently associated with COVID-19 (Dhama et al. 2020), a subset of other immune cells such as neutrophils, monocytes, and macrophages are significantly upregulated by SARS-CoV-2 infection (An et al. 2020; Huang et al. 2020; Liu et al. 2020c; Yang et al. 2020).
In general, monocytes are innate immune cells that participate in inflammatory responses, phagocytosis, antigen presentation, and a variety of other immune processes (Jakubzick et al. 2017). Circulating monocytes extravasate into peripheral tissues to differentiate into macrophages or dendritic cells during inflammation. (Jakubzick et al. 2017). Thus, monocyte function upregulation by SARS-CoV-2 infection may contribute to the strengthening of various proinflammatory processes that are mediated by activated monocytes.
Like monocytes, neutrophils are also recruited to the site of infection via circulation and the permeabilization of endothelial membranes adjacent to the infection site (Fig. 3a). However, due to COVID-19-induced lymphopenia, COVID-19 patients are more vulnerable to a microbial superinfection. As a result, an excessive number of neutrophils will infiltrate toward virus-infected cells owing to chemokine overproduction.
It has been hypothesized that the inability to eradicate SARS-CoV-2 infection due to its antagonism of the innate immune response hyperinflates the innate immune system (Yoshikawa et al. 2010; Kaplan and Radic 2012; Birra et al. 2020). Subsequently, this causes an excessive inflammatory cytokine release to compensate for the exhausted immune system due to SARS-CoV-2-induced lymphopenia (Fathi and Rezaei 2020). Regarding pulmonary pathogenesis, cytokine overproduction after SARS-CoV-2 infection will increase the membrane permeability of the capillary walls around the infected alveoli, resulting in pulmonary edema, dyspnea, and hypoxemia (Fig. 3b) (Imai et al. 2005; Meftahi et al. 2020). Introduction of plasma fluid into the alveoli and the loss of elasticity due to reduced surfactant production by infection of type 2 pneumocytes by SARS-CoV-2 cause ARDS and ALI in COVID-19 patients (Verdecchia et al. 2020).
Cytokine storm
“Cytokine storm” was coined to describe cytokine overproduction observed during graft-versus-host disease (Ferrara et al. 1993; Clark 2007). Exessive amounts of cytokines are released into the blood, resulting in systemic inflammatory reactions. This leads to vasodilation and decreased blood pressure due to plasma loss in the entire cardiovascular system, and it is ultimately followed by an entire circulatory collapse (Allegra et al. 2020a; Coperchini et al. 2020; Ragab et al. 2020; Tufan et al. 2020). In general, proinflammatory cytokines increase the expression of cell adhesion molecules on the surface of neutrophils and endothelial cells. This promotes intercellular interactions between neutrophils and endothelial cells. Also, increasing lung endothelium permeability and reducing barrier protection attracts more neutrophils to the infection site through endothelial penetration (Fig. 3b) (Polidoro et al. 2020). This inflammatory immune response dysregulation prevents adaptive immune response activation (Manjili et al. 2020). As a consequence, these SARS-CoV-2-induced immune abnormalities can cause increased microbial infections, septic shock, and severe multiple organ dysfunction (Fig. 3c) (Yang et al. 2020).
COVID-19-induced cytokine storm resembles hemophagocytic lymphohistiocytosis (HLH; also known as macrophage activation syndrome, MAS), which has been reported in patients infected with SARS-CoV (Nicholls et al. 2003). HLH is a hyperinflammatory syndrome involving fever, cytopenias, and MOF caused by uncontrolled immune activation and excessive cytokine production (La Rosee et al. 2019). The primary HLH hallmark is the overproduction of proinflammatory cytokines such as TNF-α, IFN-γ, IL-1, IL-2, and IL-6, which are released from activated macrophages and lymphocytes after several viral infections (Ramachandran et al. 2017). Regarding SARS-CoV-2 infection, cytokine overproduction and subsequent cytokine storm induce several extrapulmonary complications (Fig. 3c). They include myocardial injury, myocarditis, acute kidney injury, impaired ion transport, acute liver injury, and gastrointestinal manifestations such as diarrhea and vomiting (Gupta et al. 2020). In particular, elevated cardiac troponin levels, which are cardiac injury biomarkers, are the most commonly reported cardiac abnormality in COVID-19 patients (Bansal 2020; Imazio et al. 2020; Tomasoni et al. 2020). Myocardial injuries such as arrhythmias, acute coronary syndrome (Akhmerov and Marban 2020), heart failure, and myocardial infarction are also frequently observed in COVID-19 patients (Bandyopadhyay et al. 2020). Therefore, the cytokine storm induced by SARS-CoV-2 infection can be regarded as a self-elicited and suicidal systemic immunological reaction that can adversely affect every key organ in the body (Lippi et al. 2020). These data show the systemic detrimental effects of SARS-CoV-2 infection on the vital functions of major organs through hyper-inflammation, and following cytokine storm, they are accountable for the relatively high mortality rate of COVID-19 patients (Gupta et al. 2020).
Prothrombic phase
Activation of thrombosis
Coagulation is a natural defense mechanism that seals off damaged blood vessels and prevents local pathogens from spreading further into the systemic circulation. However, dysregulated coagulation (thrombosis) can cause pathologically detrimental effects by obstructing blood flow, which can lead to coagulation disorders with dangerous clinical consequences. In a typical blood clotting cascade, factor X is enzymatically activated by cleavage of coagulation factors from intrinsic and extrinsic pathways and tissue factor (TF)-driven platelet activation. Activated factor X converts prothrombin into thrombin. The newly generated thrombin, in turn, converts fibrinogen into fibrin. Activated fibrin initiates the self cross-linking process together with red blood cells that ultimately generates a gel-like fibrin mesh (Fig. 4).
D-dimer derives from the degradation products of this cross-linked fibrin. Therefore, the levels of D-dimer reflect both thrombin production and activation of fibrinolysis (Miesbach 2020). Interestingly, elevated D-dimer levels, prolonged prothrombin time, and low platelet count are consistently reported in COVID-19 patients (Bikdeli et al. 2020a; Zhang et al. 2020a; Zhou et al. 2020; Miesbach and Makris 2020; Salamanna et al. 2020). Also, venous thrombotic events such as pulmonary embolism are common in COVID-19 patients (Pons et al. 2020). In particular, elevated levels of D-dimer (> 1 µg/L) are strongly associated with COVID-19 patient in-hospital death (Tang et al. 2020). Fibrin deposition in air spaces and lung parenchyma were also frequently found in coronavirus-induced ARDS patients (Whyte et al. 2020).
Regarding their pathogenic mechanisms, increased platelet aggregation and upregulation of coagulation-inducing factors such as the Von Willebrand factor (vWF), factor VIII, and plasminogen activator inhibitor-1 (PAI-1) are suggested to be causative factors of coagulopathy caused by SARS-CoV-2 infection (Magro 2020; Miesbach and Makris 2020; Tang et al. 2020). In particular, the induction of PAI-1 by SARS-CoV-2 infection seems to be a significant event in the pathogenesis of COVID-19 because PAI-1 can inhibit urokinase-type plasminogen activator (uPA) and tissue plasminogen activator (tPA) (Ekholm et al. 2009; Whyte et al. 2020). Since both uPA and tPA are essential for plasmin activation, plasmin-mediated fibrinolysis is suppressed by PAI-1 induction during SARS-CoV-2 infection.
Furthermore, there seem to be several additional factors that contribute to thrombosis during SARS-CoV-2 infection. Neutrophil extracellular traps (NETs) are extracellular fiber networks primarily composed of neutrophil DNA that bind to and kill extracellular pathogens while minimizing damage to host cells (Kaplan and Radic 2012). SARS-CoV-2 infection induces NET overproduction. This increased NET production by SARS-CoV-2 may facilitate thrombus formation (Kaplan and Radic 2012; Allegra et al. 2020b; Cicco et al. 2020) (Fig. 4). Since monocytes and macrophages also produce TF, monocyte, and macrophage upregulation by SARS-CoV-2 infection can also promote TF-initiated coagulation. In addition, a proinflammatory cytokine, TNF-α, also activates TF. In particular, IL-6 pathway activation by SARS-CoV-2 infection can induce vascular endothelial growth factor (VEGF), fibrinogen, and factor VII (Bikdeli et al. 2020b; Whyte et al. 2020). Angiotensin II also stimulates TF expression in damaged cells, increases thrombin formation, and impairs fibrinolysis (Celi et al. 2010; Miesbach 2020). As explained previously, a virus-induced chronic ACE2 deficiency results in the overactivation of AT1R receptor through the prolonged stabilization of angiotensin II. Since the increased thrombosis was one of the physiologically relevant events that were triggered by the continuous activation of the angiotensin II signaling pathway (Fig. 2b), ACE2 deficiency can further aggravate the prothrombotic state, which is already stimulated by proinflammatory conditions induced by SARS-CoV-2 infection.
Therapeutic modulation
Based on the previously explained three-step pathogenesis model, various phase-specific therapeutic strategies can be pursued to intervene in COVID-19 progression. In line with the previously proposed three-step model for COVID-19 pathogenesis, a number of therapeutic approaches against COVID-19 that are currently in clinical trials will be classified and explained with an emphasis on their mechanisms of action along with typical examples (Table 1).
Pulmonary phase
Pulmonary phase-specific therapeutics include RAS inhibitors, entry inhibitors, replication inhibitors, and protease inhibitors. RAS inhibitors are further divided into ACE inhibitors and angiotensin II receptor blockers (ARBs). They alleviate the RAS overactivation caused by ACE2 deficiency after SARS-CoV-2 infection. Entry inhibitors, replication inhibitors, and protease inhibitors exert direct antiviral activities by targeting specific steps of the viral life cycle.
RAS inhibitors
RAS inhibitors such as ACE inhibitors and angiotensin II receptor blockers (ARBs) are frequently prescribed hypertensive drugs. Their use as a COVID-19 therapeutic has raised safety concerns because RAS inhibitors increase ACE2 expression in hypertensive patients (Vaduganathan et al. 2020; Aleksova et al. 2020). In theory, ACE2 upregulation caused by RAS inhibition could lead to more efficient SARS-CoV-2 entry, which makes RAS inhibitor users more vulnerable to chronic SARS-CoV-2 infection (Aleksova et al. 2020; Wang et al. 2020b). Despite these concerns, most clinical experts advise against the abrupt withdrawal of RAS inhibitors in high-risk patients, including those who have heart failure or have had a myocardial infarction because this may result in clinical instability and adverse health outcomes (Rico-Mesa et al. 2020). Until more data are available, they advised that RAS inhibitors should be continued in patients in otherwise stable conditions who are at risk for COVID-19 (Vaduganathan et al. 2020).
In general, RAS inhibitors may be beneficial to COVID-19 patients because they downregulate RAS pathways, which are overactivated by SARS-CoV-2 infection, as previously explained. Also, ACE inhibitors suppressed TMPRSS2 expression, which is an essential co-receptor for SARS-CoV-2 cell entry (Luo et al. 2020). This could help reduce new SARS-CoV-2 infections by disrupting the enzymatic digestion of the spike protein by TMPRSS2, which is necessary for membrane fusion. ACE2-mediated signaling stimulation via ACE2 gene delivery along with treatment with angiotensin 1–7 and MasR agonists was suggested as another therapeutic approach to counteract hyperactivated RAS signaling caused by ACE2 deficiency after SARS-CoV-2 infection (Verdecchia et al. 2020; Gheblawi et al. 2020).
Entry inhibitors
Considering that ACE2 plays a central role in SARS-CoV-2 entry, SARS-CoV-2 neutralization via recombinant ACE2 protein administration has been proposed as a therapeutic modality (Crackower et al. 2002). Preliminary data suggest that the surface molecule CD147 may be an alternative pathway for the virus to enter the host cells since the spike protein of SARS-CoV-2 can bind to CD147 (Gubernatorova et al. 2020). Based on these data, the efficacy of humanized antibodies against CD147 such as meplazumab is being tested in clinical trials as a COVID-19 treatment (Drozdzal et al. 2020). Also, the beneficial effects of the macrolide antibiotic azithromycin in reducing the viral load of hospitalized patients were associated with its ability to interfere with ligand/CD147 receptor interactions (Ulrich and Pillat 2020). In addition to its direct antiviral activity, azithromycin can also influence immune parameters such as increasing antiviral interferon expression in both healthy and virus-infected cells and exerting antiviral and anti-inflammatory actions (Schijns and Lavelle 2020). These potential dual mechanisms might qualify azithromycin as a promising therapeutic candidate to be tested in the future.
Replication and protease inhibitors
Replication inhibitors suppress viral RNA genome replication by antagonizing the enzymatic activity of SARS-CoV-2 RNA-dependent RNA polymerase (RDRP). For this reason, they are called RDRP inhibitors. They include remdesivir, favipiravir, and ribavirin. Remdesivir and favipiravir were originally developed to treat the Ebola virus and influenza virus, respectively (Reina and Reina 2017; Tchesnokov et al. 2019; Shiraki and Daikoku 2020; Pardo et al. 2020). Structurally, they are nucleotide analogs that hinder the functions of endogenous nucleotides as building blocks during viral RNA synthesis. Due to their potent antiviral activity toward SARS-CoV-2, they were repurposed as a SARS-CoV-2 treatment (Cao et al. 2020; Singh et al. 2020; Pardo et al. 2020; Azevedo et al. 2020; Hashemian et al. 2020; Frediansyah et al. 2020). Remdesivir was the first drug clinically approved for the treatment of COVID-19 patients. Host protease inhibitors such as nafamostat and camostat mesylate inhibit the host TMPRSS-2 protease, which is essential for SARS-CoV-2 entry into target cells (Asakura and Ogawa 2020; Hoffmann et al. 2020; Doi et al. 2020; Hifumi et al. 2020). Therefore, these host protease inhibitors likely block virus entry. Lopinavir/ritonavir belongs to another class of virus protease inhibitors. Although it was originally developed as an anti-HIV drug, it was also repurposed due to its inhibitory activity against the coronavirus protease (Stower 2020; Doggrell 2020; Osborne et al. 2020). It should be noted that ritonavir is a pharmacokinetic booster that prevents lopinavir degradation by host cytochrome enzymes.
Proinflammatory phase
Proinflammatory phase-specific therapeutics include general inflammatory drugs, cytokine inhibitors, JAK-STAT signaling inhibitors, complement pathway inhibitors, immunomodulatory drugs, cell-based therapy, and convalescent plasma therapy. General inflammatory drugs are further divided into corticosteroids and non-steroidal anti-inflammatory drugs (NSAIDs). Cytokine inhibitors target cytokines induced by SARS-CoV-2 infection. They include IL-6, IL-1β, GM-CSF, IFN-γ, TNF-α, and VEGF.
General anti-inflammatory drugs
Corticosteroids are a class of steroid hormones with anti-inflammatory and immunosuppressive activities. They are naturally produced in the adrenal cortex of vertebrates. Synthetic analogs include dexamethasone, ciclesonide, budesonide, and prednisone. They exert anti-inflammatory effects by blocking inflammatory mediator actions and inducing anti-inflammatory mediators. Their direct action on T-lymphocytes and subsequent suppression of delayed hypersensitivity reactions seem to be related to their immunosuppressive effects.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are frequently used to manage the early clinical symptoms of SARS-CoV-2 infection. The clinical efficacy of several corticosteroids and NSAIDs to slow down the progression of the pro-inflammatory phase of COVID-19 pathogenesis is being tested in numerous clinical trials worldwide (Hossein-Khannazer et al. 2020; Saghazadeh and Rezaei 2020; Zhang et al. 2020b; Ye et al. 2020).
Cytokine inhibitors
IL-6 is regarded as one of the most critical factors in the proinflammatory progression of COVID-19 pathogenesis. IL-6 was also recommended as a cytokine storm early indicator in COVID-19 patients (An et al. 2020). Therefore, antibody-based IL-6 inhibitors such as tocilizumab, sarilumab, and siltuximab were tested as pharmacological agents to reduce IL-6-mediated COVID-19 (Franco et al. 2020; Paniri and Akhavan-Niaki 2020; Gubernatorova et al. 2020; Liu et al. 2020a). Mechanistically, tocilizumab and sarilumab bind to IL-6 receptors, and siltuximab binds to IL-6, itself. As explained previously, IL-1β, GM-CSF, IFN-γ, TNF-α, and VEGF also play critical roles in COVID-19 pathogenesis by stimulating various immune responses. Therefore, IL-1β inhibitors such as canakinumab and anakinra (van de Veerdonk and Netea 2020, Alijotas-Reig et al. 2020); GM-CSF inhibitors such as mavrilimumab (Lang et al. 2020; Gremese et al. 2020); IFN-γ inhibitors such as emapalumab (Magro 2020); TNF-α inhibitors such as infliximab, adalimumab, golimumab, certolizumab, and etanercept (Soy et al. 2020); and VEGF inhibitors such as bevacizumab and ranibizumab (Polidoro et al. 2020)were suggested as potential COVID-19 drugs.
JAK inhibitors such as baricitinib, ruxolitinib, and tofacitinib are frequently used to treat cancers induced by JAK-STAT pathway overactivation. The JAK-STAT pathway is critical in delivering IFN, cytokine, and chemokine signals to target cells. Therefore, its inhibition by JAK inhibitors is effective against the COVID-19 proinflammatory stage pathogenesis progression induced by IFN, cytokine, and chemokine overproduction (Colafrancesco et al. 2020; Alijotas-Reig et al. 2020; Zhang et al. 2020b; Luo et al. 2020).
A complementary pathway inhibitor such as C5-targeting eculizumab was also predicted to alleviate complementary activation by SARS-CoV-2 infection (Magro 2020). The use of immunomodulatory drugs such as a sphingosine-1 phosphate receptor regulator like Fingolimod (FTY720) was also suggested for the treatment of COVID-19 (Barzegar et al. 2020; Foerch et al. 2020).
Classic antimalarial drugs such as chloroquine and hydroxychloroquine also exhibited anti-SARS-CoV-2 activity by two mechanistically distinct activities. Their immunomodulatory properties seem to reduce the secretion of important proinflammatory cytokines such as TNF-α and IL-6 by peripheral blood mononuclear cells (van den Borne et al. 1997). Their ability to halt the progression from early endosome to lysosome also seems to decrease the viral replication rate (Liu et al. 2020b).
Lastly, a cell-based therapy that uses mesenchymal stem cells with immunomodulatory properties and a convalescent plasma therapy are in clinical trials to test their efficacy in alleviating COVID-19 pathogenesis progression (Wang et al. 2016).
Prothrombic phase
Prothrombic phase-specific therapeutics include vitamin K antagonists, antiplatelets, and anti-Xa agents. Mechanistically, vitamin K epoxide reductase, ADP receptor, and thrombin inhibition as well as antithrombin activation suppressed COVID-19-induced coagulopathy .
Vitamin K antagonists
Warfarin is commonly used as an oral anticoagulant to treat coagulation disorders such as deep vein thrombosis and pulmonary embolism. It is also used to prevent stroke in people with heart disease. Warfarin decreases blood clotting by blocking an enzyme called vitamin K epoxide reductase. This enzyme helps reactivate vitamin K1, upon which clotting factors such as II, VII, IX, and X depend.
Antiplatelets
Adenosine diphosphate (ADP) receptor inhibitors are antiplatelet agents. They include aspirin, ticagrelor, prasugrel, clopidogrel, and dipyridamole. They are used to prevent coagulation-initiated events such as thromboembolism, myocardial infarction, and stroke. These drugs inhibit ADP from binding to the P2Y12 receptor on platelets, which inhibits platelet aggregation and thrombus formation.
Anti-Xa agents
In general, anti-Xa agents are classified into heparin-related products and direct thrombin inhibitors. Heparin is a naturally occurring polysaccharide. It can activate the enzyme inhibitor antithrombin III (AT) by inducing a conformational change. The heparin-activated AT then inactivates thrombin, factor Xa, and other proteases that are necessary for blood clot formation (Mitchell 2020). Unfractionated heparin and low molecular-weight heparin are in clinical trials to test whether their anticoagulation effects delay COVID-19 prothrombic phase pathogenesis progression (Maldonado et al. 2020; Bikdeli et al. 2020b). Direct thrombin inhibitors are another class of anticoagulants that directly inhibit the enzyme thrombin. They include danaparoid, fondaparinux, bivalirudin, dabigatran, argatroban, apixaban, and rivaroxaban.
Conclusion and perspective
As shown in the three-step COVID-19 pathogenesis model, SARS-CoV-2 infection produces a dynamic spectrum of clinical manifestations based on complicated host and viral responses. Therefore, COVID-19 pathogenesis stage-specific therapeutics should be selected to maximize patient recovery and survival. Early therapeutic approaches should target the virus-specific or host-virus interaction steps in the virus life cycle. Early approaches should also focus on restoring RAS balance by stimulating ACE2-mediated signaling. Therapeutic strategies targeting middle to late-stage pathogenesis should focus more on alleviating exaggerated host responses to the viral infection such as cytokine overproduction, systemic inflammation, dysregulated platelet aggregation, and coagulopathy. More basic research and clinical studies will be required to find ideal drug combinations with multiple mechanisms of action against stage-specific COVID-19 pathogenesis.

COVID-19 pathogenesis model. The three phases of COVID-19 pathogenesis: pulmonary, proinflammatory, and prothrombic. Response patterns, pathogenic mechanisms, and clinical manifestations for each phase are described

Pulmonary phase of COVID-19 pathogenesis. a SARS-CoV-2 enters the type 2 pneumocyte via the host receptor, ACE2. Target cell infection by SARS-CoV-2 induces ACE2 internalization, resulting in ACE2 downregulation and deficiency. b SARS-CoV-2-induced ACE2 deficiency decreases the conversion of angiotensin II to angiotensinogen 1–7 and increases angiotensin II availability. Excessive angiotensin II causes AT1R overactivation, resulting in a RAS imbalance. c ADAM-17 activation by AT1R promotes cleavage of both membrane-anchored TNF-α and ACE2. ADAM-17 truncates the ACE2 extracellular domain, which enzymatically inactivates ACE2. As a result, des-Arg9-BK degradation by ACE2 is disrupted and its abundancy increases. Liberated soluble TNF-α and accumulated des-Arg9-BK aggravate SARS-CoV-2-induced inflammation


Pro-inflammatory phase of COVID-19 pathogenesis. a After target cell entry, innate receptors such as RIG-I, TLRs, and MDA5 recognize SARS-CoV-2 RNA motifs. The NLRP3 inflammasome recognizes DAMPs, which are generated by SARS-CoV-2 infection. This virus-receptor recognition activates transcription factors such as IRFs and NF-kB, activating the transcription of target genes such as IFNs, cytokines, and chemokines. The secretion of these proteins induces endothelial activation by increasing blood vessel permeability. This allows leukocytes to infiltrate the site of infection. b SARS-CoV-2 infection of type 2 pneumocytes leads to cytokine overproduction. This increases the membrane permeability of the capillary walls around the infected alveoli, resulting in pulmonary edema. Due to this pulmonary edema, proper gas exchange is impaired in COVID-19 patients. This is clinically manifested by dyspnea, hypoxemia, and ARDS. c Cytokine overproduction and cytokine storm induce clinically relevant extrapulmonary effects on various key organs such as the heart, kidney, liver, and intestines

Prothrombic phase of COVID-19 pathogenesis. SARS-CoV-2 infection induces RAS overactivation, NETs formation, cytokine overproduction, and hyperinflammation. These effects translate into increased risk for coagulation disorders
References
Akhmerov A, Marban E (2020) COVID-19 and the Heart. Circ Res 126:1443–1455. doi:https://doi.org/10.1161/CIRCRESAHA.120.317055
Aleksova A, Ferro F, Gagno G, Cappelletto C, Santon D, Rossi M, Ippolito G, Zumla A, Beltrami AP, Sinagra G (2020) COVID-19 and renin-angiotensin system inhibition: role of angiotensin converting enzyme 2 (ACE2)—is there any scientific evidence for controversy? J Intern Med 288:410–421. https://doi.org/10.1111/joim.13101
Alijotas-Reig J, Esteve-Valverde E, Belizna C, Selva-O’callaghan A, Pardos-Gea J, Quintana A, Mekinian A, Anunciacion-Llunell A, Miro-Mur F (2020) Immunomodulatory therapy for the management of severe COVID-19. Beyond the anti-viral therapy: a comprehensive review. Autoimmun Rev 19:102569. https://doi.org/10.1016/j.autrev.2020.102569
Allegra A, Di Gioacchino M, Tonacci A, Musolino C, Gangemi S (2020) Immunopathology of SARS-CoV-2 infection: immune cells and mediators, prognostic factors, and immune-therapeutic implications. Int J Mol Sci 21:4782. https://doi.org/10.3390/ijms21134782
Allegra A, Innao V, Allegra AG, Musolino C (2020) Coagulopathy and thromboembolic events in patients with SARS-CoV-2 infection: pathogenesis and management strategies. Ann Hematol 99(9):1953–1965. https://doi.org/10.1007/s00277-020-04182-4
An PJ, Zhu YZ, Yang LP (2020) Biochemical indicators of coronavirus disease 2019 exacerbation and the clinical implications. Pharmacol Res 159:104946. https://doi.org/10.1016/j.phrs.2020.104946
Asakura H, Ogawa H (2020) Potential of heparin and nafamostat combination therapy for COVID-19. J Thromb Haemost 18:1521–1522. doi:https://doi.org/10.1111/jth.14858
Azevedo TCP, Azevedo PCP, Silveira Filho RN, Carvalho A, Cezarotti Filho ML, Barbosa FT, Sousa-Rodrigues CF, Matos-Rocha TJ, Ramos F (2020) Use of remdesivir for patients with Covid-19: a review article. Rev Assoc Med Bras 66:838–841. doi:https://doi.org/10.1590/1806-9282.66.6.838
Bandyopadhyay D, Akhtar T, Hajra A, Gupta M, Das A, Chakraborty S, Pal I, Patel N, Amgai B, Ghosh RK, Fonarow GC, Lavie CJ, Naidu SS (2020) COVID-19 pandemic: cardiovascular complications and future implications. Am J Cardiovasc Drugs 20(4):311–324. https://doi.org/10.1007/s40256-020-00420-2
Bansal M (2020) Cardiovascular disease and COVID-19. Diabetes Metab Syndr 14:247–250. doi:https://doi.org/10.1016/j.dsx.2020.03.013
Banu N, Panikar SS, Leal LR, Leal AR (2020) Protective role of ACE2 and its downregulation in SARS-CoV-2 infection leading to Macrophage Activation Syndrome: therapeutic implications. Life Sci 256:117905. https://doi.org/10.1016/j.lfs.2020.117905
Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL (2013) Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123::3025–3036. doi:https://doi.org/10.1172/JCI68782
Barzegar M, Mirmosayyeb O, Nehzat N, Sarrafi R, Khorvash F, Maghzi AH, Shaygannejad V (2020) COVID-19 infection in a patient with multiple sclerosis treated with fingolimod. Neurol Neuroimmunol Neuroinflamm 7(4):e753. https://doi.org/10.1212/NXI.0000000000000753
Bikdeli B, Madhavan MV, Gupta A, Jimenez D, Burton JR, Der Nigoghossian C, Chuich T, Nouri SN, Dreyfus I, Driggin E, Sethi S, Sehgal K, Chatterjee S, Ageno W, Madjid M, Guo Y, Tang LV, Hu Y, Bertoletti L, Giri J, Cushman M, Quere I, Dimakakos EP, Gibson CM, Lippi G, Favaloro EJ, Fareed J, Tafur AJ, Francese DP, Batra J, Falanga A, Clerkin KJ, Uriel N, Kirtane A, Mclintock C, Hunt BJ, Spyropoulos AC, Barnes GD, Eikelboom JW, Weinberg I, Schulman S, Carrier M, Piazza G, Beckman JA, Leon MB, Stone GW, Rosenkranz S, Goldhaber SZ, Parikh SA, Monreal M, Krumholz HM, Konstantinides SV, Weitz JI, Lip GYH, Global C-TCG (2020) Pharmacological agents targeting thromboinflammation in COVID-19: review and implications for future research. Thromb Haemost 120:1004–1024. https://doi.org/10.1055/s-0040-1713152
Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, Nigoghossian C, Ageno W, Madjid M, Guo Y, Tang LV, Hu Y, Giri J, Cushman M, Quere I, Dimakakos EP, Gibson CM, Lippi G, Favaloro EJ, Fareed J, Caprini JA, Tafur AJ, Burton JR, Francese DP, Wang EY, Falanga A, Mclintock C, Hunt BJ, Spyropoulos AC, Barnes GD, Eikelboom JW, Weinberg I, Schulman S, Carrier M, Piazza G, Beckman JA, Steg PG, Stone GW, Rosenkranz S, Goldhaber SZ, Parikh SA, Monreal M, Krumholz HM, Konstantinides SV, Weitz JI, Lip GYH, Global COVID-19 Thrombosis Collaborative Group, Endorsed by the ISTH, NATF, ESVM, and the IUA, Supported by the ESC Working Group on Pulmonary Circulation and Right Ventricular Function (2020) COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up: JACC state-of-the-art review. J Am Coll Cardiol 75:2950–2973. https://doi.org/10.1016/j.jacc.2020.04.031
Birra D, Benucci M, Landolfi L, Merchionda A, Loi G, Amato P, Licata G, Quartuccio L, Triggiani M, Moscato P (2020) COVID 19: a clue from innate immunity. Immunol Res 68:161–168. doi:https://doi.org/10.1007/s12026-020-09137-5
Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP (1997) A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385:729–733. https://doi.org/10.1038/385729a0
Cao YC, Deng QX, Dai SX (2020) Remdesivir for severe acute respiratory syndrome coronavirus 2 causing COVID-19: an evaluation of the evidence. Travel Med Infect Dis 35:101647. https://doi.org/10.1016/j.tmaid.2020.101647
Celi A, Cianchetti S, Dell’omo G, Pedrinelli R (2010) Angiotensin II, tissue factor and the thrombotic paradox of hypertension. Expert Rev Cardiovasc Ther 8::1723–1729. doi:https://doi.org/10.1586/erc.10.161
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S (2016) Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19:181–193. https://doi.org/10.1016/j.chom.2016.01.007
Channappanavar R, Perlman S (2017) Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol 39:529–539. doi:https://doi.org/10.1007/s00281-017-0629-x
Chen IY, Moriyama M, Chang MF, Ichinohe T (2019) Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol 10:50. https://doi.org/10.3389/fmicb.2019.00050
Chen J, Subbarao K (2007) The Immunobiology of SARS*. Annu Rev Immunol 25:443–472. doi:https://doi.org/10.1146/annurev.immunol.25.022106.141706
Cicco S, Cicco G, Racanelli V, Vacca A (2020) Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): two potential targets for COVID-19 treatment. Mediators Inflamm 2020:7527953. https://doi.org/10.1155/2020/7527953
Clark IA (2007) The advent of the cytokine storm. Immunol Cell Biol 85:271–273. doi:https://doi.org/10.1038/sj.icb.7100062
Colafrancesco S, Scrivo R, Barbati C, Conti F, Priori R (2020) Targeting the immune system for pulmonary inflammation and cardiovascular complications in COVID-19 patients. Front Immunol 11:1439. https://doi.org/10.3389/fimmu.2020.01439
Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M (2020) The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev 53:25–32. https://doi.org/10.1016/j.cytogfr.2020.05.003
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-Dos-Santos AJ, Da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM (2002) Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417:822–828. doi:https://doi.org/10.1038/nature00786
Dhama K, Patel SK, Pathak M, Yatoo MI, Tiwari R, Malik YS, Singh R, Sah R, Rabaan AA, Bonilla-Aldana DK, Rodriguez-Morales AJ (2020) An update on SARS-CoV-2/COVID-19 with particular reference to its clinical pathology, pathogenesis, immunopathology and mitigation strategies. Travel Med Infect Dis 37:101755. doi:https://doi.org/10.1016/j.tmaid.2020.101755
Dhochak N, Singhal T, Kabra SK, Lodha R (2020) Pathophysiology of COVID-19: why children fare better than adults? Indian J Pediatr 87:537–546. https://doi.org/10.1007/s12098-020-03322-y
Doggrell SA (2020) Does lopinavir measure up in the treatment of COVID-19? Expert Opin Investig Drugs 29(8):793–796. https://doi.org/10.1080/13543784.2020.1777277
Doi K, Ikeda M, Hayase N, Moriya K, Morimura N, Group C-US (2020) Nafamostat mesylate treatment in combination with favipiravir for patients critically ill with Covid-19: a case series. Crit Care 24::392. doi:https://doi.org/10.1186/s13054-020-03078-z
Domingo P, Mur I, Pomar V, Corominas H, Casademont J, De Benito N (2020) The four horsemen of a viral apocalypse: the pathogenesis of SARS-CoV-2 infection (COVID-19). EBioMedicine 58:102887. https://doi.org/10.1016/j.ebiom.2020.102887
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 87::E1–E9. doi:https://doi.org/10.1161/01.res.87.5.e1
Drozdzal S, Rosik J, Lechowicz K, Machaj F, Kotfis K, Ghavami S, Los MJ (2020) FDA approved drugs with pharmacotherapeutic potential for SARS-CoV-2 (COVID-19) therapy. Drug Resist Updat 53::100719. doi:https://doi.org/10.1016/j.drup.2020.100719
Ekholm M, Kahan T, Jorneskog G, Broijersen A, Wallen NH (2009) Angiotensin II infusion in man is proinflammatory but has no short-term effects on thrombin generation in vivo. Thromb Res 124::110–115. doi:https://doi.org/10.1016/j.thromres.2008.12.040
Fathi N, Rezaei N (2020) Lymphopenia in COVID-19: therapeutic opportunities. Cell Biol Int. https://doi.org/10.1002/cbin.11403
Ferrara JL, Abhyankar S, Gilliland DG (1993) Cytokine storm of graft-versus-host disease: a critical effector role for interleukin-1. Transplant Proc 25:1216–1217
Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73:1907–1916. https://doi.org/10.1128/IAI.73.4.1907-1916.2005
Foerch C, Friedauer L, Bauer B, Wolf T, Adam EH (2020) Severe COVID-19 infection in a patient with multiple sclerosis treated with fingolimod. Mult Scler Relat Disord 42:102180. https://doi.org/10.1016/j.msard.2020.102180
Franco R, Rivas-Santisteban R, Serrano-Marin J, Rodriguez-Perez AI, Labandeira-Garcia JL, Navarro G (2020) SARS-CoV-2 as a factor to disbalance the renin-angiotensin system: a suspect in the case of exacerbated IL-6 production. J Immunol. https://doi.org/10.4049/jimmunol.2000642
Frediansyah A, Nainu F, Dhama K, Mudatsir M, Harapan H (2020) Remdesivir and its antiviral activity against COVID-19: a systematic review. Clin Epidemiol Glob Health. https://doi.org/10.1016/j.cegh.2020.07.011
Gao YM, Xu G, Wang B, Liu BC (2020) Cytokine storm syndrome in coronavirus disease 2019: a narrative review. J Intern Med. https://doi.org/10.1111/joim.13144
Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, Raizada MK, Grant MB, Oudit GY (2020) Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res 126:1456–1474. https://doi.org/10.1161/CIRCRESAHA.120.317015
Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, Simmons G, Hofmann H, Kuri T, Weber F, Eichler J, Drosten C, Pohlmann S (2010) Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol 84:1198–1205. doi:https://doi.org/10.1128/JVI.01248-09
Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pohlmann S (2011) Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol 85:4122–4134. doi:https://doi.org/10.1128/JVI.02232-10
Gremese E, Ferraccioli ES, Alivernini S, Tolusso B, Ferraccioli G (2020) Basic immunology may lead to translational therapeutic rationale: SARS-CoV-2 and rheumatic diseases. Eur J Clin Invest 50(9):e13342. https://doi.org/10.1111/eci.13342
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Zhan J, Wang S, Xie Z, Zhuang H, Wu B, Zhong H, Shao H, Fang W, Gao D, Pei F, Li X, He Z, Xu D, Shi X, Anderson VM, Leong AS (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202:415–424. doi:https://doi.org/10.1084/jem.20050828
Gubernatorova EO, Gorshkova EA, Polinova AI, Drutskaya MS (2020) IL-6: relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev 53:13–24. https://doi.org/10.1016/j.cytogfr.2020.05.009
Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, Bikdeli B, Ahluwalia N, Ausiello JC, Wan EY, Freedberg DE, Kirtane AJ, Parikh SA, Maurer MS, Nordvig AS, Accili D, Bathon JM, Mohan S, Bauer KA, Leon MB, Krumholz HM, Uriel N, Mehra MR, Elkind MSV, Stone GW, Schwartz A, Ho DD, Bilezikian JP, Landry DW (2020) Extrapulmonary manifestations of COVID-19. Nat Med 26:1017–1032. https://doi.org/10.1038/s41591-020-0968-3
Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, Yamamoto N, Sasazuki T, Ishizaka Y (2008) Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci USA 105:7809–7814. doi:https://doi.org/10.1073/pnas.0711241105
Hashemian SM, Farhadi T, Velayati AA (2020) A review on remdesivir: a possible promising agent for the treatment of COVID-19. Drug Des Dev Ther 14:3215–3222. https://doi.org/10.2147/DDDT.S261154
Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G (2020) Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta 507:167–173. https://doi.org/10.1016/j.cca.2020.04.027
Hifumi T, Isokawa S, Otani N, Ishimatsu S (2020) Adverse events associated with nafamostat mesylate and favipiravir treatment in COVID-19 patients. Crit Care 24::497. doi:https://doi.org/10.1186/s13054-020-03227-4
Hoffmann M, Schroeder S, Kleine-Weber H, Muller MA, Drosten C, Pohlmann S (2020) Nafamostat mesylate blocks activation of SARS-CoV-2: new treatment option for COVID-19. Antimicrob Agents Chemother 64(6):e00754-20. https://doi.org/10.1128/AAC.00754-20
Hossein-Khannazer N, Shokoohian B, Shpichka A, Aghdaei HA, Timashev P, Vosough M (2020) Novel therapeutic approaches for treatment of COVID-19. J Mol Med (Berl) 98:789–803. doi:https://doi.org/10.1007/s00109-020-01927-6
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506. doi:https://doi.org/10.1016/S0140-6736(20)30183-5
Imai Y, Kuba K, Penninger JM (2008) The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol 93:543–548. doi:https://doi.org/10.1113/expphysiol.2007.040048
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436:112–116. doi:https://doi.org/10.1038/nature03712
Imazio M, Klingel K, Kindermann I, Brucato A, De Rosa FG, Adler Y, De Ferrari GM (2020) COVID-19 pandemic and troponin: indirect myocardial injury, myocardial inflammation or myocarditis? Heart 106:1127–1131. doi:https://doi.org/10.1136/heartjnl-2020-317186
Jakubzick CV, Randolph GJ, Henson PM (2017) Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol 17:349–362. doi:https://doi.org/10.1038/nri.2017.28
Jiang F, Yang J, Zhang Y, Dong M, Wang S, Zhang Q, Liu FF, Zhang K, Zhang C (2014) Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat Rev Cardiol 11:413–426. doi:https://doi.org/10.1038/nrcardio.2014.59
Kaplan MJ, Radic M (2012) Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 189:2689–2695. doi:https://doi.org/10.4049/jimmunol.1201719
Kowalinski E, Lunardi T, Mccarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S (2011) Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147:423–435. doi:https://doi.org/10.1016/j.cell.2011.09.039
Kuba K, Imai Y, Penninger JM (2006) Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol 6::271–276. doi:https://doi.org/10.1016/j.coph.2006.03.001
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11:875–879. doi:https://doi.org/10.1038/nm1267
La Rosee P, Horne A, Hines M, Von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J, Girschikofsky M, Jordan MB, Kumar A, Van Laar JaM, Lachmann G, Nichols KE, Ramanan AV, Wang Y, Wang Z, Janka G, Henter JI (2019) Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 133:2465–2477. doi:https://doi.org/10.1182/blood.2018894618
Lang FM, Lee KM, Teijaro JR, Becher B, Hamilton JA (2020) GM-CSF-based treatments in COVID-19: reconciling opposing therapeutic approaches. Nat Rev Immunol 20:507–514. doi:https://doi.org/10.1038/s41577-020-0357-7
Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, Pan P, Wang W, Hu D, Liu X, Zhang Q, Wu J (2020) Coronavirus infections and immune responses. J Med Virol 92:424–432. doi:https://doi.org/10.1002/jmv.25685
Lippi G, Sanchis-Gomar F, Henry BM (2020) COVID-19: unravelling the clinical progression of nature’s virtually perfect biological weapon. Ann Transl Med 8::693. doi:https://doi.org/10.21037/atm-20-3989
Liu B, Li M, Zhou Z, Guan X, Xiang Y (2020a) Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun 111::102452. doi:https://doi.org/10.1016/j.jaut.2020.102452
Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, Li Y, Hu Z, Zhong W, Wang M (2020b) Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov 6::16. doi:https://doi.org/10.1038/s41421-020-0156-0
Liu X, Cao H, Li J, Wang B, Zhang P, Dong Zhang X, Liu Z, Yuan H, Zhan Z (2017) Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ 24:683–693. doi:https://doi.org/10.1038/cdd.2017.1
Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, Wang Z, Li J, Li J, Feng C, Zhang Z, Wang L, Peng L, Chen L, Qin Y, Zhao D, Tan S, Yin L, Xu J, Zhou C, Jiang C, Liu L (2020c) Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci 63:364–374. doi:https://doi.org/10.1007/s11427-020-1643-8
Luo W, Li YX, Jiang LJ, Chen Q, Wang T, Ye DW (2020) Targeting JAK-STAT signaling to control cytokine release syndrome in COVID-19. Trends Pharmacol Sci 41:531–543. https://doi.org/10.1016/j.tips.2020.06.007
Magro G (2020) COVID-19: review on latest available drugs and therapies against SARS-CoV-2. Coagulation and inflammation cross-talking. Virus Res 286:198070. https://doi.org/10.1016/j.virusres.2020.198070
Mahmudpour M, Roozbeh J, Keshavarz M, Farrokhi S, Nabipour I (2020) COVID-19 cytokine storm: the anger of inflammation. Cytokine 133:155151. https://doi.org/10.1016/j.cyto.2020.155151
Maldonado E, Tao D, Mackey K (2020) Antithrombotic therapies in COVID-19 disease: a systematic review. J Gen Intern Med 35:2698. https://doi.org/10.1007/s11606-020-05906-y
Manjili RH, Zarei M, Habibi M, Manjili MH (2020) COVID-19 as an acute inflammatory disease. J Immunol 205:12–19. https://doi.org/10.4049/jimmunol.2000413
Market M, Angka L, Martel AB, Bastin D, Olanubi O, Tennakoon G, Boucher DM, Ng J, Ardolino M, Auer RC (2020) Flattening the COVID-19 curve with natural killer cell based immunotherapies. Front Immunol 11:1512. https://doi.org/10.3389/fimmu.2020.01512
Meftahi GH, Jangravi Z, Sahraei H, Bahari Z (2020) The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: the contribution of “inflame-aging”. Inflamm Res 69::825–839. doi:https://doi.org/10.1007/s00011-020-01372-8
Miesbach W (2020) Pathological role of angiotensin II in severe COVID-19. TH Open 4:e138–e144. https://doi.org/10.1055/s-0040-1713678
Miesbach W, Makris M (2020) COVID-19: coagulopathy, risk of thrombosis, and the rationale for anticoagulation. Clin Appl Thromb Hemost 26:1076029620938149. https://doi.org/10.1177/1076029620938149
Mitchell WB (2020) Thromboinflammation in COVID-19 acute lung injury. Paediatr Respir Rev. doi:https://doi.org/10.1016/j.prrv.2020.06.004
Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, Mccauley P, Mcgeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD, Et Al (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385:733–736 doi:https://doi.org/10.1038/385733a0
Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, Chu CM, Hui PK, Mak KL, Lim W, Yan KW, Chan KH, Tsang NC, Guan Y, Yuen KY, Peiris JS (2003) Lung pathology of fatal severe acute respiratory syndrome. Lancet 361:1773–1778. doi:https://doi.org/10.1016/s0140-6736(03)13413-7
Nikonov A, Molder T, Sikut R, Kiiver K, Mannik A, Toots U, Lulla A, Lulla V, Utt A, Merits A, Ustav M (2013) RIG-I and MDA-5 detection of viral RNA-dependent RNA polymerase activity restricts positive-strand RNA virus replication. PLoS Pathog 9:e1003610. doi:https://doi.org/10.1371/journal.ppat.1003610
Osborne V, Davies M, Lane S, Evans A, Denyer J, Dhanda S, Roy D, Shakir S (2020) Lopinavir-ritonavir in the treatment of COVID-19: a dynamic systematic benefit-risk assessment. Drug Saf 43:809–821. https://doi.org/10.1007/s40264-020-00966-9
Paces J, Strizova Z, Smrz D, Cerny J (2020) COVID-19 and the immune system. Physiol Res 69(3):379-388. https://doi.org/10.33549/physiolres.934492
Paniri A, Akhavan-Niaki H (2020) Emerging role of IL-6 and NLRP3 inflammasome as potential therapeutic targets to combat COVID-19: role of lncRNAs in cytokine storm modulation. Life Sci 257:118114. https://doi.org/10.1016/j.lfs.2020.118114
Pardo J, Shukla AM, Chamarthi G, Gupte A (2020) The journey of remdesivir: from Ebola to COVID-19. Drugs Context. https://doi.org/10.7573/dic.2020-4-14
Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, Putko B, Kassiri Z, Turner AJ, Oudit GY (2014) Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol 66::167–176. doi:https://doi.org/10.1016/j.yjmcc.2013.11.017
Patel VB, Mori J, Mclean BA, Basu R, Das SK, Ramprasath T, Parajuli N, Penninger JM, Grant MB, Lopaschuk GD, Oudit GY (2016) ACE2 deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes 65:85–95. https://doi.org/10.2337/db15-0399
Picchianti Diamanti A, Rosado MM, Pioli C, Sesti G, Lagana B (2020) Cytokine release syndrome in COVID-19 patients, a new scenario for an old concern: the fragile balance between infections and autoimmunity. Int J Mol Sci. doi:https://doi.org/10.3390/ijms21093330
Polidoro RB, Hagan RS, De Santis Santiago R, Schmidt NW (2020) Overview: Systemic inflammatory response derived from lung injury caused by SARS-CoV-2 infection explains severe outcomes in COVID-19. Front Immunol 11:1626. https://doi.org/10.3389/fimmu.2020.01626
Pons S, Fodil S, Azoulay E, Zafrani L (2020) The vascular endothelium: the cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit Care 24::353. doi:https://doi.org/10.1186/s13054-020-03062-7
Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R (2020) The COVID-19 cytokine storm; what we know so far. Front Immunol 11:1446. https://doi.org/10.3389/fimmu.2020.01446
Ramachandran S, Zaidi F, Aggarwal A, Gera R (2017) Recent advances in diagnostic and therapeutic guidelines for primary and secondary hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis 64:53–57. doi:https://doi.org/10.1016/j.bcmd.2016.10.023
Reina J, Reina N (2017) Favipiravir, a new concept of antiviral drug against influenza viruses. Rev Esp Quimioter 30:79–83
Rico-Mesa JS, White A, Anderson AS (2020) Outcomes in patients with COVID-19 infection taking ACEI/ARB. Curr Cardiol Rep 22:31. doi:https://doi.org/10.1007/s11886-020-01291-4
Saghazadeh A, Rezaei N (2020) Towards treatment planning of COVID-19: rationale and hypothesis for the use of multiple immunosuppressive agents: anti-antibodies, immunoglobulins, and corticosteroids. Int Immunopharmacol 84:106560. https://doi.org/10.1016/j.intimp.2020.106560
Salamanna F, Maglio M, Landini MP, Fini M (2020) Platelet functions and activities as potential hematologic parameters related to Coronavirus Disease 2019 (Covid-19). Platelets 31:627–632. doi:https://doi.org/10.1080/09537104.2020.1762852
Sallenave JM, Guillot L (2020) Innate immune signaling and proteolytic pathways in the resolution or exacerbation of SARS-CoV-2 in Covid-19: key therapeutic targets? Front Immunol 11:1229. https://doi.org/10.3389/fimmu.2020.01229
Scala S, Pacelli R (2020) Fighting the host reaction to SARS-COv-2 in critically ill patients: the possible contribution of off-label drugs. Front Immunol 11:1201. https://doi.org/10.3389/fimmu.2020.01201
Schijns V, Lavelle EC (2020) Prevention and treatment of COVID-19 disease by controlled modulation of innate immunity. Eur J Immunol 50:932–938. doi:https://doi.org/10.1002/eji.202048693
Shi CS, Nabar NR, Huang NN, Kehrl JH (2019) SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov 5:101. https://doi.org/10.1038/s41420-019-0181-7
Shiraki K, Daikoku T (2020) Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol Ther 209::107512. doi:https://doi.org/10.1016/j.pharmthera.2020.107512
Singh AK, Singh A, Singh R, Misra A (2020) Remdesivir in COVID-19: a critical review of pharmacology, pre-clinical and clinical studies. Diabetes Metab Syndr 14:641–648. https://doi.org/10.1016/j.dsx.2020.05.018
Soy M, Keser G, Atagunduz P, Tabak F, Atagunduz I, Kayhan S (2020) Cytokine storm in COVID-19: pathogenesis and overview of anti-inflammatory agents used in treatment. Clin Rheumatol 39::2085–2094. doi:https://doi.org/10.1007/s10067-020-05190-5
Stower H (2020) Lopinavir-ritonavir in severe COVID-19. Nat Med 26::465. doi:https://doi.org/10.1038/s41591-020-0849-9
Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT (2012) PAMPs and DAMPs: signal 0 s that spur autophagy and immunity. Immunol Rev 249:158–175. doi:https://doi.org/10.1111/j.1600-065X.2012.01146.x
Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18:844–847. doi:https://doi.org/10.1111/jth.14768
Tchesnokov EP, Feng JY, Porter DP, Gotte M (2019) Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses 11:326. https://doi.org/10.3390/v11040326
Tomasoni D, Italia L, Adamo M, Inciardi RM, Lombardi CM, Solomon SD, Metra M (2020) COVID-19 and heart failure: from infection to inflammation and angiotensin II stimulation. Searching for evidence from a new disease. Eur J Heart Fail 22:957–966. doi:https://doi.org/10.1002/ejhf.1871
Tseng YH, Yang RC, Lu TS (2020) Two hits to the renin-angiotensin system may play a key role in severe COVID-19. Kaohsiung J Med Sci 36::389–392. doi:https://doi.org/10.1002/kjm2.12237
Tufan A, Avanoglu Guler A, Matucci-Cerinic M (2020) COVID-19, immune system response, hyperinflammation and repurposing antirheumatic drugs. Turk J Med Sci 50:620–632. doi:https://doi.org/10.3906/sag-2004-168
Turner AJ, Hiscox JA, Hooper NM (2004) ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci 25::291–294. doi:https://doi.org/10.1016/j.tips.2004.04.001
Ulrich H, Pillat MM (2020) CD147 as a target for COVID-19 treatment: suggested effects of azithromycin and stem cell engagement. Stem Cell Rev Rep 16:434–440. https://doi.org/10.1007/s12015-020-09976-7
Vaduganathan M, Vardeny O, Michel T, Mcmurray JJV, Pfeffer MA, Solomon SD (2020) Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med 382:1653–1659. https://doi.org/10.1056/NEJMsr2005760
Van De Veerdonk FL, Netea MG (2020) Blocking IL-1 to prevent respiratory failure in COVID-19. Crit Care 24:445. doi:https://doi.org/10.1186/s13054-020-03166-0
Van Den Borne BE, Dijkmans BA, De Rooij HH, Le Cessie S, Verweij CL (1997) Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J Rheumatol 24:55–60
Verdecchia P, Cavallini C, Spanevello A, Angeli F (2020) The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med 76:14–20. doi:https://doi.org/10.1016/j.ejim.2020.04.037
Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P (2002) Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 277::14838–14843. doi:https://doi.org/10.1074/jbc.M200581200
Wang J, Jiang M, Chen X, Montaner LJ (2020) Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol 108:17–41. https://doi.org/10.1002/JLB.3COVR0520-272R
Wang JJ, Edin ML, Zeldin DC, Li C, Wang DW, Chen C (2020b) Good or bad: application of RAAS inhibitors in COVID-19 patients with cardiovascular comorbidities. Pharmacol Ther. https://doi.org/10.1016/j.pharmthera.2020.107628
Wang K, Gheblawi M, Oudit GY (2020) Angiotensin converting enzyme 2: a double-edged sword. Circulation 142:426–428. https://doi.org/10.1161/CIRCULATIONAHA.120.047049
Wang LT, Ting CH, Yen ML, Liu KJ, Sytwu HK, Wu KK, Yen BL (2016) Human mesenchymal stem cells (MSCs) for treatment towards immune- and inflammation-mediated diseases: review of current clinical trials. J Biomed Sci 23:76. https://doi.org/10.1186/s12929-016-0289-5
Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ (2020) Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost 18:1548–1555. doi:https://doi.org/10.1111/jth.14872
Yang L, Liu S, Liu J, Zhang Z, Wan X, Huang B, Chen Y, Zhang Y (2020) COVID-19: immunopathogenesis and Immunotherapeutics. Signal Transduct Target Ther 5::128. doi:https://doi.org/10.1038/s41392-020-00243-2
Ye Q, Wang B, Mao J (2020) The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect 80:607–613. doi:https://doi.org/10.1016/j.jinf.2020.03.037
Yoshikawa T, Hill TE, Yoshikawa N, Popov VL, Galindo CL, Garner HR, Peters CJ, Tseng CT (2010) Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome-associated coronavirus infection. PLoS ONE 5:e8729. https://doi.org/10.1371/journal.pone.0008729
Zhang JJ, Dong X, Cao YY, Yuan YD, Yang YB, Yan YQ, Akdis CA, Gao YD (2020a) Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 75:1730–1741. doi:https://doi.org/10.1111/all.14238
Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, Wang J, Qin Y, Zhang X, Yan X, Zeng X, Zhang S (2020) The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. Clin Immunol 214:108393. https://doi.org/10.1016/j.clim.2020.108393
Zhang YH, Zhang YH, Dong XF, Hao QQ, Zhou XM, Yu QT, Li SY, Chen X, Tengbeh AF, Dong B, Zhang Y (2015) ACE2 and Ang-(1–7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm Res 64:253–260. doi:https://doi.org/10.1007/s00011-015-0805-1
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395:1054–1062. https://doi.org/10.1016/S0140-6736(20)30566-3
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education, Science, and Technology (NRF-2019R1F1A1058628). Choongho Lee generated all scientific illustrations shown in the figures by using the web drawing service provided by biorender.com. We would like to thank Amina Tucak for providing the original template on which Fig. 3b is based.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lee, C., Choi, W.J. Overview of COVID-19 inflammatory pathogenesis from the therapeutic perspective. Arch. Pharm. Res. 44, 99–116 (2021). https://doi.org/10.1007/s12272-020-01301-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-020-01301-7
Keywords
- Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
- Coronavirus disease 2019 (COVID-19)
- Renin-angiotensin system (RAS)
- Angiotensin-converting enzyme 2 (ACE2)
- ACE2 deficiency
- Acute respiratory distress syndrome (ARDS)
- Acute lung injury (ALI)
- Cytokine storm
- Thrombosis
- Coagulopathy
- Multi-organ failure (MOF)