
Abstract
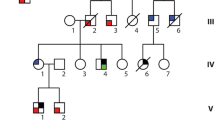
Dilated cardiomyopathy (DCM) is the most prevalent cause of non-ischemic cardiac failure and the commonest indication for cardiac transplantation. Compelling evidence highlights the pivotal roles of genetic defects in the occurrence of DCM. Nevertheless, the genetic determinants underpinning DCM remain largely obscure. In this study, the coding regions of ISL1, which encodes a transcription factor critical for embryonic cardiogenesis and postnatal cardiac remodeling, were sequenced in 216 unrelated patients with DCM, and a novel heterozygous ISL1 mutation, NM_002202.2: c.631A>T; p.(Lys211*), was identified in a proband. The mutation, which co-segregated with DCM in the family, was absent in 238 unrelated controls, as well as in the Genome Aggregation and the Exome Aggregation Consortium population databases. Functional analyses unveiled that the mutant ISL1 protein lost transcriptional activity alone or in synergy with TBX20 or GATA4, two other transcription factors associated with DCM. These findings indicate ISL1 as a new gene of DCM.




Similar content being viewed by others
Abbreviations
- DCM:
-
Dilated cardiomyopathy
- CHD:
-
Congenital heart disease
- VSD:
-
Ventricular septal defect
- PCR:
-
Polymerase chain reaction
- SD:
-
Standard deviation
- HD:
-
Homeodomain
- TAD:
-
Transcriptional activation domain
References
Merlo, M., Cannatà, A., Gobbo, M., Stolfo, D., Elliott, P. M., & Sinagra, G. (2018). Evolving concepts in dilated cardiomyopathy. European Journal of Heart Failure, 20, 228–239.
Tabish, A. M., Azzimato, V., Alexiadis, A., Buyandelger, B., & Knöll, R. (2017). Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophysical Reviews, 9, 207–223.
Halliday, B. P., Gulati, A., Ali, A., Guha, K., Newsome, S., Arzanauskaite, M., et al. (2017). Association between midwall late gadolinium enhancement and sudden cardiac death in patients with dilated cardiomyopathy and mild and moderate left ventricular systolic dysfunction. Circulation, 135, 2106–2115.
Halliday, B. P., Cleland, J. G. F., Goldberger, J. J., & Prasad, S. K. (2017). Personalizing risk stratification for sudden death in dilated cardiomyopathy: the past, present, and future. Circulation, 136, 215–231.
Stolfo, D., Ceschia, N., Zecchin, M., De Luca, A., Gobbo, M., Barbati, G., et al. (2018). Arrhythmic risk stratification in patients with idiopathic dilated cardiomyopathy. The American Journal of Cardiology, 121, 1601–1609.
Norum, H. M., Broch, K., Michelsen, A. E., Lunde, I. G., Lekva, T., Abraityte, A., et al. (2017). The notch ligands DLL1 and periostin are associated with symptom severity and diastolic function in dilated cardiomyopathy. Journal of Cardiovascular Translational Research, 10, 401–410.
McNally, E. M., & Mestroni, L. (2017). Dilated cardiomyopathy: genetic determinants and mechanisms. Circulation Research, 121, 731–748.
Weintraub, R. G., Semsarian, C., & Macdonald, P. (2017). Dilated cardiomyopathy. Lancet, 390, 400–414.
Lee, Y. Z. J., & Judge, D. P. (2017). The role of genetics in peripartum cardiomyopathy. Journal of Cardiovascular Translational Research, 10, 437–445.
Bondue, A., Arbustini, E., Bianco, A., Ciccarelli, M., Dawson, D., De Rosa, M., et al. (2018). Complex roads from genotype to phenotype in dilated cardiomyopathy: scientific update from the working Group of Myocardial Function of the European Society of Cardiology. Cardiovascular Research, 114, 1287–1303.
Shakeel, M., Irfan, M., & Khan, I. A. (2018). Rare genetic mutations in Pakistani patients with dilated cardiomyopathy. Gene, 673, 134–139.
Iuso, A., Wiersma, M., Schüller, H. J., Pode-Shakked, B., Marek-Yagel, D., Grigat, M., et al. (2018). Mutations in PPCS, encoding phosphopantothenoylcysteine synthetase, cause autosomal-recessive dilated cardiomyopathy. American Journal of Human Genetics, 102, 1018–1030.
Nozari, A., Aghaei-Moghadam, E., Zeinaloo, A., Mollazadeh, R., Majnoon, M. T., Alavi, A., et al. (2018). A novel splicing variant in FLNC gene responsible for a highly penetrant familial dilated cardiomyopathy in an extended Iranian family. Gene, 659, 160–167.
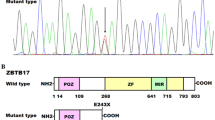
Sun, Y. M., Wang, J., Xu, Y. J., Wang, X. H., Yuan, F., Liu, H., et al. (2018). ZBTB17 loss-of-function mutation contributes to familial dilated cardiomyopathy. Heart and Vessels, 33, 722–732.
Barefield, D. Y., Puckelwartz, M. J., Kim, E. Y., Wilsbacher, L. D., Vo, A. H., Waters, E. A., et al. (2017). Experimental modeling supports a role for MyBP-HL as a novel myofilament component in arrhythmia and dilated cardiomyopathy. Circulation, 136, 1477–1491.
Tayal, U., Newsome, S., Buchan, R., Whiffin, N., Walsh, R., Barton, P. J., et al. (2017). Truncating variants in titin independently predict early arrhythmias in patients with dilated cardiomyopathy. Journal of the American College of Cardiology, 68, 2466–2468.
Long, P. A., Theis, J. L., Shih, Y. H., Maleszewski, J. J., Abell Aleff, P. C., Evans, J. M., et al. (2017). Recessive TAF1A mutations reveal ribosomopathy in siblings with end-stage pediatric dilated cardiomyopathy. Human Molecular Genetics, 26, 2874–2881.
Cao, S., Smith, L. L., Padilla-Lopez, S. R., Guida, B. S., Blume, E., Shi, J., et al. (2017). Homozygous EEF1A2 mutation causes dilated cardiomyopathy, failure to thrive, global developmental delay, epilepsy and early death. Human Molecular Genetics, 26, 3545–3552.
Zhou, C., Li, C., Zhou, B., Sun, H., Koullourou, V., Holt, I., et al. (2017). Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Human Molecular Genetics, 26, 2258–2276.
Jansweijer, J. A., Nieuwhof, K., Russo, F., Hoorntje, E. T., Jongbloed, J. D., Lekanne Deprez, R. H., et al. (2017). Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. European Journal of Heart Failure, 19, 512–521.
Le Dour, C., Macquart, C., Sera, F., Homma, S., Bonne, G., Morrow, J. P., et al. (2017). Decreased WNT/β-catenin signaling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin A/C gene. Human Molecular Genetics, 26, 333–343.
Dal Ferro, M., Stolfo, D., Altinier, A., Gigli, M., Perrieri, M., Ramani, F., et al. (2017). Association between mutation status and left ventricular reverse remodeling in dilated cardiomyopathy. Heart, 103, 1704–1710.
Janin, A., N'Guyen, K., Habib, G., Dauphin, C., Chanavat, V., Bouvagnet, P., et al. (2017). Truncating mutations on myofibrillar myopathies causing genes as prevalent molecular explanations on patients with dilated cardiomyopathy. Clinical Genetics, 92, 616–623.
Robyns, T., Kuiperi, C., Breckpot, J., Devriendt, K., Souche, E., Van Cleemput, J., et al. (2017). Repeat genetic testing with targeted capture sequencing in primary arrhythmia syndrome and cardiomyopathy. European Journal of Human Genetics, 25, 1313–1323.
Petropoulou, E., Soltani, M., Firoozabadi, A. D., Namayandeh, S. M., Crockford, J., Maroofian, R., et al. (2017). Digenic inheritance of mutations in the cardiac troponin (TNNT2) and cardiac beta myosin heavy chain (MYH7) as the cause of severe dilated cardiomyopathy. European Journal of Medical Genetics, 60, 485–488.
Zaidi, S., & Brueckner, M. (2017). Genetics and genomics of congenital heart disease. Circulation Research, 120, 923–940.
Blue, G. M., Kirk, E. P., Giannoulatou, E., Sholler, G. F., Dunwoodie, S. L., Harvey, R. P., et al. (2017). Advances in the genetics of congenital heart disease: a clinician's guide. Journal of the American College of Cardiology, 69, 859–870.
Li, Y. J., & Yang, Y. Q. (2017). An update on the molecular diagnosis of congenital heart disease: focus on loss-of-function mutations. Expert Review of Molecular Diagnostics, 17, 393–401.
Li, R. G., Li, L., Qiu, X. B., Yuan, F., Xu, L., Li, X., et al. (2013). GATA4 loss-of-function mutation underlies familial dilated cardiomyopathy. Biochemical and Biophysical Research Communications, 439, 591–596.
Zhang, X. L., Dai, N., Tang, K., Chen, Y. Q., Chen, W., Wang, J., et al. (2015). GATA5 loss-of-function mutation in familial dilated cardiomyopathy. International Journal of Molecular Medicine, 35, 763–770.
Xu, L., Zhao, L., Yuan, F., Jiang, W. F., Liu, H., Li, R. G., et al. (2014). GATA6 loss-of-function mutations contribute to familial dilated cardiomyopathy. International Journal of Molecular Medicine, 34, 1315–1322.
Zhang, X. L., Qiu, X. B., Yuan, F., Wang, J., Zhao, C. M., Li, R. G., et al. (2015). TBX5 loss-of-function mutation contributes to familial dilated cardiomyopathy. Biochemical and Biophysical Research Communications, 459, 166–171.
Kirk, E. P., Sunde, M., Costa, M. W., Rankin, S. A., Wolstein, O., Castro, M. L., et al. (2007). Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. American Journal of Human Genetics, 81, 280–291.
Costa, M. W., Guo, G., Wolstein, O., Vale, M., Castro, M. L., Wang, L., et al. (2013). Functional characterization of a novel mutation in NKX2-5 associated with congenital heart disease and adult-onset cardiomyopathy. Circulation Cardiovascular Genetics, 6, 238–247.
Yuan, F., Qiu, Z. H., Wang, X. H., Sun, Y. M., Wang, J., Li, R. G., et al. (2018). MEF2C loss-of-function mutation associated with familial dilated cardiomyopathy. Clinical Chemistry and Laboratory Medicine, 56, 502–511.
Zhou, Y. M., Dai, X. Y., Qiu, X. B., Yuan, F., Li, R. G., Xu, Y. J., et al. (2016). HAND1 loss-of-function mutation associated with familial dilated cardiomyopathy. Clinical Chemistry and Laboratory Medicine, 54, 1161–1167.
Qiu, X. B., Qu, X. K., Li, R. G., Liu, H., Xu, Y. J., Zhang, M., et al. (2017). CASZ1 loss-of-function mutation contributes to familial dilated cardiomyopathy. Clinical Chemistry and Laboratory Medicine, 55, 1417–1425.
Cai, C. L., Liang, X., Shi, Y., Chu, P. H., Pfaff, S. L., Chen, J., et al. (2003). Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Developmental Cell, 5, 877–889.
Laugwitz, K. L., Moretti, A., Lam, J., Gruber, P., Chen, Y., Woodard, S., et al. (2005). Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature, 433, 647–653.
Moretti, A., Caron, L., Nakano, A., Lam, J. T., Bernshausen, A., Chen, Y., et al. (2006). Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell, 127, 1151–1165.
Bu, L., Jiang, X., Martin-Puig, S., Caron, L., Zhu, S., Shao, Y., et al. (2009). Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature, 460, 113–117.
Osoegawa, K., Schultz, K., Yun, K., Mohammed, N., Shaw, G. M., & Lammer, E. J. (2014). Haploinsufficiency of insulin gene enhancer protein 1 (ISL1) is associated with d-transposition of the great arteries. Molecular Genetics and Genomic Medicine, 2, 341–351.
Takeuchi, J. K., Mileikovskaia, M., Koshiba-Takeuchi, K., Heidt, A. B., Mori, A. D., Arruda, E. P., et al. (2005). Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development, 132, 2463–2474.
Stadiotti, I., Catto, V., Casella, M., Tondo, C., Pompilio, G., & Sommariva, E. (2017). Arrhythmogenic cardiomyopathy: the guilty party in adipogenesis. Journal of Cardiovascular Translational Research, 10, 446–454.
Zhang, M., Chen, J., Si, D., Zheng, Y., Jiao, H., Feng, Z., et al. (2014). Whole exome sequencing identifies a novel EMD mutation in a Chinese family with dilated cardiomyopathy. BMC Medical Genetics, 15, 77.
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genetics in Medicine, 17, 405–424.
Dodou, E., Verzi, M. P., Anderson, J. P., Xu, S. M., & Black, B. L. (2004). Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development, 131, 3931–3942.
Brown, C. O., 3rd, Chi, X., Garcia-Gras, E., Shirai, M., Feng, X. H., & Schwartz, R. J. (2004). The cardiac determination factor, Nkx2-5, is activated by mutual cofactors GATA-4 and Smad1/4 via a novel upstream enhancer. The Journal of Biological Chemistry, 279, 10659–10669.
Zhao, C. M., Sun, B., Song, H. M., Wang, J., Xu, W. J., Jiang, J. F., et al. (2016). TBX20 loss-of-function mutation associated with familial dilated cardiomyopathy. Clinical Chemistry and Laboratory Medicine, 54, 325–332.
Friedrich, F. W., Dilanian, G., Khattar, P., Juhr, D., Gueneau, L., Charron, P., et al. (2013). A novel genetic variant in the transcription factor Islet-1 exerts gain of function on myocyte enhancer factor 2C promoter activity. European Journal of Heart Failure, 15, 267–276.
Moretti, A., Bellin, M., Jung, C. B., Thies, T. M., Takashima, Y., Bernshausen, A., et al. (2010). Mouse and human induced pluripotent stem cells as a source for multipotent Isl1+ cardiovascular progenitors. Federation of American Societies for Experimental Biology Journal, 24, 700–711.
Liang, X., Zhang, Q., Cattaneo, P., Zhuang, S., Gong, X., Spann, N. J., et al. (2015). Transcription factor ISL1 is essential for pacemaker development and function. The Journal of Clinical Investigation, 125, 3256–3268.
Genead, R., Danielsson, C., Andersson, A. B., Corbascio, M., Franco-Cereceda, A., Sylven, C., et al. (2010). Islet-1 cells are cardiac progenitors present during the entire lifespan: from the embryonic stage to adulthood. Stem Cells and Development, 19, 1601–1615.
Li, Y., Tian, S., Lei, I., Liu, L., Ma, P., & Wang, Z. (2017). Transplantation of multipotent Isl1+ cardiac progenitor cells preserves infarcted heart function in mice. American Journal of Translational Research, 9, 1530–1542.
Wang, L., Meier, E. M., Tian, S., Lei, I., Liu, L., Xian, S., et al. (2017). Transplantation of Isl1+ cardiac progenitor cells in small intestinal submucosa improves infarcted heart function. Stem Cell Research and Therapy, 8, 230.
Liu, H., Xu, Y. J., Li, R. G., Wang, Z. S., Zhang, M., Qu, X. K., et al. (2018). HAND2 loss-of-function mutation causes familial dilated cardiomyopathy. European Journal of Medical Genetics. https://doi.org/10.1016/j.ejmg.2018.09.007.
Stevens, K. N., Hakonarson, H., Kim, C. E., Doevendans, P. A., Koeleman, B. P., Mital, S., et al. (2010). Common variation in ISL1 confers genetic susceptibility for human congenital heart disease. PLoS One, 5, e10855.
Acknowledgments
The authors acknowledge the study subjects for their participation in the study.
Funding
This work was financially supported by the grants from the National Natural Science Foundation of China (No. 81470372), the Natural Science Foundation of Minhang District, Shanghai, China (No. 2018MHZ072), the Program of the Health and Family Planning Commission of Shanghai, China (No. M20170348), and the Key Project of the Fifth People’s Hospital of Shanghai, Fudan University, China (No. 2018WYZD05).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was carried out in conformity with the ethical standards established in the 1964 Declaration of Helsinki and its later amendments and was approved by the Medical Ethics Committee of the Fifth People′s Hospital of Shanghai, Fudan University, China. All study participants provided their informed consent prior to the study.
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. No animal studies were carried out by the authors for this article.
Informed Consent
Informed consent was obtained from all study subjects.
Additional information
Associate Editor Paul J. R. Barton oversaw the review of this article
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, YJ., Wang, ZS., Yang, CX. et al. Identification and Functional Characterization of an ISL1 Mutation Predisposing to Dilated Cardiomyopathy. J. of Cardiovasc. Trans. Res. 12, 257–267 (2019). https://doi.org/10.1007/s12265-018-9851-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-018-9851-8