
Abstract
Long-term fructose consumption has been shown to evoke leptin resistance, to elevate triglyceride levels and to induce insulin resistance and hepatic steatosis. Autophagy has been suggested to function in processes such as lipid storage in adipose tissue and inflammation in liver. Autophagy and the leptin system have also been suggested to regulate each other. This study aimed to identify the changes caused by fetal undernourishment and postnatal fructose diet in the gene expression of leptin, its receptors (LEPR-a, LEPR-b, LEPR-c, LEPR-e and LEPR-f) and autophagy genes in the white adipose tissue (WAT) and liver of adult male rats in order to clarify the mechanism behind the metabolic alterations. The data clearly revealed that the long-term postnatal fructose diet decreased leptin levels (p < 0.001), LEPR (p < 0.001), especially LEPR-b (p = 0.011) and LEPR-f (p = 0.005), as well as SOCS3 (p < 0.001), ACC (p = 0.006), ATG7 (p < 0.001), MAP1LC3β (p < 0.001) and LAMP2 (p = 0.004) mRNA expression in WAT. Furthermore, LEPR (p < 0.001), especially LEPR-b (p = 0.001) and LEPR-f (p < 0.001), ACC (p = 0.010), ATG7 (p = 0.024), MAP1LC3β (p = 0.003) and LAMP2 (p < 0.001) mRNA expression in the liver was increased in fructose-fed rats. In addition, the LEPR expression in liver and MAP1LC3β expression in WAT together explained 55.7 % of the variation in the plasma triglyceride levels of the rats (R 2adj. = 0.557, p < 0.001). These results, together with increased p62 levels in WAT (p < 0.001), could indicate decreased adipose tissue lipid storing capacity as well as alterations in liver metabolism which may represent a plausible mechanism through which fructose consumption could disturb lipid metabolism and result in elevated triglyceride levels.
Similar content being viewed by others


Introduction
Previous studies have indicated that the individual risk of developing metabolic syndrome and related disorders could be determined already during the fetal development. Particularly nutrition in early life can act to program the fetal metabolism, and this can be the basis for diseases encountered in adult life (McMillen and Robinson 2005). Fetal growth restriction has been associated with the development of the features of the metabolic syndrome, including dyslipidemia, visceral obesity and impaired glucose tolerance (Briana and Malamitsi-Puchner 2009). We have recently shown that intra-uterine growth restriction (IUGR) led to a decreased weight and increased plasma LDL cholesterol levels but higher insulin sensitivity in adult male rats (Malo et al. 2013). Dietary exposure to fructose has recently increased, and there is a concern that excessive fructose consumption in humans may be one of the factors behind the rising incidence of obesity worldwide. Fructose is a highly lipogenic sugar that has been associated with several components of the metabolic syndrome such as insulin resistance, elevated waist circumference, dyslipidemia and hypertension. A high-fructose diet has also been shown to induce leptin resistance (Dekker et al. 2010). In our previous study, male rats consuming 60 % fructose diet were smaller, had higher plasma triglycerides (TGs) and fasting insulin levels, decreased plasma LDL cholesterol, impaired glucose tolerance and a higher lipid content in their retroperitoneal and intra-abdominal adipose tissue (Malo et al. 2013).
Adipose tissue is suggested to play an important role in linking the poor fetal growth and the later development of adult diseases (Jaquet et al. 2005). Leptin, a hormone expressed by the Lep gene mainly in adipocytes, controls food intake and energy expenditure (Zhang et al. 1994). The effects of leptin are mediated through its receptor (LEPR), which is not only located in the central nervous system but also in other tissues, including adipocytes (Tartaglia et al. 1995; Zhang et al. 1994). Suppressor of cytokine signaling 3 (SOCS3) gene expression has been demonstrated to be induced by leptin, and it has been shown to act as a negative regulator of leptin signaling (Bjorbaek et al. 1999). Adipocyte-specific deficiency of leptin receptors has been shown to result in increased adiposity, decreased energy expenditure, insulin resistance and dyslipidemia (Huan et al. 2003). Many of these features are also seen as a result of IUGR and fructose consumption. On the other hand, overexpression of LEPR-b in adipose tissue on mice has been found to lead to elevated TG levels, ectopic fat accumulation and hyperinsulinemia in mice (Wang et al. 2008).
In adipocytes, autophagy has been proposed to play a role in adipogenesis (Singh et al. 2009b; Zhang et al. 2009). Autophagy-related gene 7 (ATG7) encodes an enzyme that is essential for autophagosome formation. ATG7 knockout mice exhibited impaired autophagy which lead to decreased lipid storage in WAT (Singh et al. 2009b; Zhang et al. 2009) and lower leptin secretion from adipocytes (Zhang et al. 2009). Moreover, autophagy has been shown to be upregulated in the adipose tissue of obese human subjects and inhibition of autophagy results in increased production of proinflammatory cytokines but unchanged leptin levels (Jansen et al. 2012). In the liver, the role of autophagy is less clear, but it may be involved in the lipid droplet formation (Shibata et al. 2009) or lipid breakdown (lipophagy; Singh et al. 2009a). It has also been demonstrated that leptin induces autophagy in peripheral tissues including liver (Malik et al. 2011).
Here, we studied the effect of IUGR and a postnatal fructose diet on the leptin system and autophagy genes ATG7, microtubule-associated protein 1 light chain 3 beta (MAP1LC3β) and lysosomal-associated membrane protein 2 (LAMP2) expression and investigated if these changes could explain the metabolic changes encountered in IUGR and fructose consumption. In addition, we measured the LC3 protein levels in WAT and liver and investigated LC3-II, the cleaved and lipid bound form of LC3 as well as p62 protein levels, to assess the amount of autophagy. We also examined microscopically the size of adipocytes in visceral adipose tissue and the extent of fat accumulation in liver as well as the gene expression of acetyl-coA carboxylase alpha (ACC) in both tissues.
Methods
Animals
Sprague–Dawley rats were obtained from the Center of Experimental Animals of the University of Oulu, Finland. The experimental design was approved by the Animal Care and Use Committee of the University of Oulu, Finland. Nine-week-old first-time pregnant rats were randomly assigned into two dietary treatment groups on day four of gestation. A group of control dams was fed ad libitum with standard laboratory chow (Lactamin R36; energy content 3.1 kcal/g, 18.5 % protein and 4.0 % fat) throughout the pregnancy. The group of food-restricted dams received 50 % of ad libitum food intake, which had been determined earlier (Hietaniemi et al. 2009). All dams were housed individually and had free access to water during 12 h/12 h light/dark cycles. After delivery, all dams and offspring received food ad libitum.
The day of parturition was defined as postnatal day 0. On postnatal day 1, the pups were weighed and the litter size was adjusted to eight pups per litter: four males and four females. In this study, two males per litter were included. Half of the pups delivered by food-restricted dams (R) were randomly cross-fostered to control dams (C), while the rest of the pups were nursed by their own dams. Thus, the following study groups were assigned: ad libitum fed dams with their own pups (CC), ad libitum fed dams with pups born from food-restricted dams (RC) and food-restricted dams with their own pups (RR).
From 1 month of age onwards, half of the litters of each study group received a fructose-rich diet (fru) (Harlan Teklad TD89247 60 % fructose diet, energy density 3.6 kcal/g, 20.2 % of calories from protein, 12.9 % from fat and 66.8 % from carbohydrate) and the other half continued to receive the standard diet (st). The study design is shown in Fig. 1. At the age of 6 months, six standard-diet-fed litters from groups CC and RC and seven litters of RR as well as six fructose-diet-fed litters from each group were studied as described in the following. Two male offspring from each litter were fasted for 12 h, anesthetized by isoflurane inhalation and killed by decapitation. Blood was collected via cardiac puncture into heparinized vacutainers and centrifuged. Plasma was removed and stored at −70 °C for later use. The white adipose tissue and liver samples were quickly collected, immediately flash-frozen in liquid nitrogen and stored at −70 °C.

The study design. C = ad libitum fed dam, R = 50 % food-restricted dam, CC = offspring of C dam, RR = offspring of R dam lactated by its own mother and RC = offspring delivered by R dam and cross-fostered to C dam. Abbreviations st and fru refer to standard and fructose diets, respectively, starting from the age of 1 month
Microscopical analysis of the maximal adipocyte size
The size of adipocytes was microscopically determined from visceral adipose tissue samples. The samples from 5 to 6 male rats/group were prepared as paraffin blocks and stained with hematoxylin and eosin. The slides were photographed at 20× magnification using Leitz Aristoplasn microscope connected to MIcroPublisher 5.0 RTV digital camera and computer. We used MCID-Image analysis software to objectively analyze the adipocyte sizes. Certain threshold for cell color was carefully adjusted. From every individual, there were two slides of which two fields were photographed. From every field, the five largest adipocytes were selected and analyzed with MCID Core software (version 7.0). The maximal adipocytes were selected to minimize the variation due to different cut levels of the cells: by selection of maximal diameter, we wanted to avoid the cells which were cut at the edges. The mean of the twenty adipocyte maximal area per rat was calculated. The analysis was performed double-blinded, i.e., the person preparing the slides had no information about the study design, and at this point, the slides were given new identifier numbers for microscopy; thus, the person performing this was ignorant of the study groups.
Microscopical analysis of the liver fat content
The liver samples from 6 to 7 male rats/group were prepared as paraffin blocks and stained with hematoxylin and eosin. The slides were studied at 63x magnification using Leica DM3000 microscope connected to digital camera Leica DEC420 and computer and photographed with LAS V4.1.0 software. From every rat, there was one slide of which 20 fields were examined. A test grid of 100 points was used for every field, and for each point, it was defined if it was in cytoplasm or lipid vacuole. The ratio was calculated by dividing the vacuole points with the sum of cytoplasmic and vacuole points. The analysis was performed double-blinded as the person preparing the slides had no information about the study design, and at this point, the slides were given new identifier numbers for microscopy; thus, the person performing this was unaware of the study groups.
Plasma measurements
Plasma was obtained from blood collected from heart as a terminal puncture into heparinized vacutainers, centrifuged and supernatant stored at −70 °C. Leptin was measured from plasma samples with a commercial Rat Leptin ELISA Kit (Millipore). The analyses of plasma TG as well as fasting insulin and glucose measurements have been previously described in detail (Malo et al. 2013).
Total RNA extraction
The total RNA was extracted from the tissues collected from the pups at the age of 1 and 6 months. The tissues were homogenized with TissueLyser LT (Qiagen) according to the custom protocol defined in manufacturer’s instructions. The RNA was extracted from adipose tissue with RNeasy Lipid Tissue Mini Kit (Qiagen) and from liver with RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocols. The RNA concentration and purity were determined spectrophotometrically at wavelengths 260 and 280 nm.
Synthesis of the cDNA
The mRNA was converted to cDNA with RevertAid First Strand cDNA Synthesis Kit (Fermentas) using 1 μg of total RNA and Oligo (dT)18 primers supplied in a kit. Prior to the synthesis, the genomic DNA was removed from RNA preparation with DNase treatment (Fermentas). Both protocols were performed by following the instructions of the manufacturer.
Quantitative real-time PCR
The gene expression was studied by using quantitative real-time PCR (qRT-PCR). This was performed for 12 pups from each group except for 14 pups from RRst group. The measurements were done with SYBR Green chemistry with an iQ5 real-time PCR detection system (Bio-Rad). The PCR program started with 3 min in 95 °C. The 40 cycles of the PCR program consisted of 10 s at 95 °C, annealing for 10 s and extension at 72 °C for 10 s. These were followed by the final step of 2 min at 72 °C. The primers for the genes and isoforms and the annealing temperatures are presented in Table 1. The expression of all LEPR isoforms relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was first determined in general with standard-diet-fed animals (n = 38) pooled as a sample and with total cDNA concentration of 0.2 μg. The isoforms with the expression on a measurable level were then studied in individual animals. These results were normalized with the gene expression of GAPDH and 18S rRNA in WAT or hypoxanthine guanine phosphoribosyl transferase (HPRT) and ubiquitin C (UBC) in liver measured from the same samples. To minimize the possible effect of study design on the reference gene expressions, the reference genes were selected from a few candidates with NormFinder software (Andersen et al. 2004).
Protein extraction
Samples from WAT were homogenized by TissueLyser (Qiagen) for 2 min in 35 MHz in lysis buffer (20 mM Tris, 100 mM NaCl, 0.4 % SDS, 0.2 % Triton-X) with protease inhibitor and centrifuged with 13,200 rpm in 4 °C for 10 min. The liver samples were treated as adipose tissue samples except no SDS included. Supernatants were quantified for protein with DC Protein Assay (Bio-Rad), measured with 650 nm and frozen in −70 °C for future use.
Western blot
The samples were treated with β-mercaptoethanol and boiled in 95 °C for 10 min; 50 mg of total protein from liver or 10 mg of WAT was loaded onto 15 % gels for studying LC3 and 30 mg of total protein of WAT onto 12 % gels for p62 study. Samples were separated by SDS-PAGE first with 80 V 10 min and then 100 V, transferred to PVDF membrane 30 V for 90 min and blocked with 10 % non-fat milk for 1 h. Membranes were incubated with primary antibody anti-LC3 (LC3-5F10; Nanotools Antikoerpertechnik) dilution of 1:200, anti-p62 (ab56416; Abcam) with a dilution of 1:667 or anti-β-actin (A5441, Sigma–Aldrich) with a dilution of 1:3,000 overnight in 4 °C and then with secondary antibody anti-mouse (NA931 V; GE Healthcare Life Sciences) dilution 1:1,500–1:3,000 for 1 h in room temperature. The secondary antibody was detected by ECL method. Fluorescent densities were determined using Quantity One software (Bio-Rad). The intensity of LC3-II and LC3-II/LC3-I ratio was calculated. Positive control cell lysate (0911/Neuro2A/Co; Nanotools Antikoerpertechnik) was used for normalizing the differences between the LC3 blots and a control sample from CCst group for p62 blots. Western blot was performed for 6–7 rats per group.
Statistical analyses
The data were analyzed with IBM SPSS Statistics for Windows, version 19.0. (Armonk, NY: IBM Corp.). Three-way analysis of variance (ANOVA) was performed to study the effect of fetal undernourishment (IUGR or normal conditions during pregnancy) and postnatal diet (fructose-rich or standard chow from 1 to 6 month of age). In addition, suckling period (lactating dam food-restricted during pregnancy or not) was included in the analysis to cover all the different treatments between the study groups. Three-way ANOVA was done to compare the plasma leptin levels and expression values of the studied genes and protein levels between the groups as well as adipocyte size and liver lipid accumulation. The protein levels were normalized with both against total protein as well as against β-actin. However, as it seems that the traditional reference genes for Western blot might be affected by fructose feeding, the final analysis of protein levels was performed relative to the total protein an β-actin is presented in the figures only. A logarithmic transformation was applied to normalize the skewed distributions. A t test was performed when appropriate and correlations were tested with Pearson’s correlation. The relationship between TG levels and gene expression was further studied by linear regression modeling using both a one-by-one approach and stepwise modeling of SPSS. Results with a p value <0.05 were considered statistically significant.
Results
The effect of fetal undernourishment was evident in the 1-day-old pups of the food-restricted dams. At the end of the pregnancy, the food-restricted dams had gained significantly less weight (mean 48.7 g; SD 20.2 g) than control dams (mean 157.3 g; SD 25.1 g; t test; p < 0.001) and were also leaner after delivery (mean 240.0 g; SD 26.6 g) than control dams (mean 299.3 g; SD 25.3 g; t test; p = 0.002). Food-restricted pups were significantly smaller at birth (p < 0.001) compared to the pups of the ad libitum fed dams as has been already reported in our previous study (Malo et al. 2013), i.e., evidence of IUGR. The litter size was not affected by the food restriction.
The adipocyte size
The size of the adipocytes was microscopically studied from visceral adipose tissue samples. The means of the maximal adipocyte size were 26,380.0 μm2 (SD 7,561.1 μm2) in CCst, 20,483.3 μm2 (SD 3,709.9 μm2) in RCst, 25,217.7 μm2 (SD 4,428.7 μm2) in RRst, 25,370.5 μm2 (SD 5,768.0 μm2) in CCfru, 22,694.5 μm2 (SD 6,920.6 μm2) in RCfru and 25,129.4 μm2 (SD 6,719.9 μm2) in RRfru (n = 5–6/group). The size of adipocytes correlated significantly with the weight at the age of 6 months (r = 0.622, p < 0.001). There were no statistical differences between the groups in maximal adipocyte size according to IUGR or fructose diet, although a trend toward smaller adipocytes in both RCst and RCfru groups was seen. The microscope images from standard-diet-fed and fructose-diet-fed CC rat adipose tissue are shown in Fig. 2.

Histological images demonstrating the adipocyte size of a standard-diet-fed (CCst) and b fructose-fed (CCfru) rat with 20× magnification and HE-staining. A trend toward decreased adipocyte size is seen in the fructose-diet-fed rats, but it fails to reach the statistical significance
Liver fat content
The liver lipid accumulation was microscopically determined. The ratio of vacuole points to vacuole and cytoplasmic points varied between standard- and fructose-diet-fed rats (three-way ANOVA; p = 0.025) with fructose-fed rats displaying more lipid vacuoles in their livers. IUGR had no impact on liver fat accumulation. The mean ratios were 0.005 (SD 0.003) in CCst, 0.002 (SD 0.003) in RCst, 0.002 (SD 0.002) in RRst, 0.012 (SD 0.011) in CCfru, 0.007 (SD 0.006) in RCfru and 0.016 (SD 0.013) in RRfru (n = 6–7/group). The microscope images from the liver of standard-diet-fed and fructose-fed rats are shown in Fig. 3.

Histological images demonstrating the lipid content of the liver of a standard-diet-fed (CCst) and b fructose-fed (CCfru) rat with 40x magnification and HE-staining. The increased fat accumulation as lipid droplets is seen in the liver of fructose-diet-fed rat
LEPR isoform mRNA expression in adipose tissue and liver of standard-diet-fed rats
The expressions of the LEPR isoforms a, b and f were found to be at a measurable level with LEPR-f being interestingly the most abundant in both WAT and liver (Fig. 4) in the pooled sample of standard-diet-fed rats. The expression of these isoforms was further studied by qRT-PCR (n = 12–14/group). The specific mRNA expression changes induced by fructose diet, and IUGR are described in the next chapters.

Expression of LEPR isoforms in a WAT and b liver of 6-month-old male rats. Analysis is performed as a pooled sample of the standard-diet-fed rats (CCst, RCst and RRst). The isoform expression is calculated in relation to GAPDH
Effects of long-term fructose diet
Fructose did not seem to affect the plasma leptin levels significantly, although leptin mRNA expression was clearly downregulated by fructose consumption (three-way ANOVA; p < 0.001; Fig. 5). General LEPR expression was significantly downregulated in the WAT of fructose-fed animals (three-way ANOVA; p < 0.001) as shown in Fig. 6a. In particular, LEPR-b and LEPR-f mRNA levels had declined in WAT as a result of fructose consumption (three-way ANOVA; p = 0.011 and p = 0.005) together with the strongly decreased SOCS3 expression (three-way ANOVA; p < 0.001). The WAT expression of ACC mRNA was also decreased (three-way ANOVA; p = 0.006) in fructose-diet-fed rats. Furthermore, autophagy genes, ATG7 and LAMP2, were downregulated in WAT (three-way ANOVA; p < 0.001 and p = 0.004) as shown in Fig. 7a–c. The expression of MAP1LC3β mRNA was similarly decreased (three-way ANOVA; p < 0.001) in WAT. However, in the Western blot, only LC3-II band was detected in WAT and no statistically significant differences were seen in the protein levels (three-way ANOVA; p = 0.065; Fig. 8a–c). Also p62 protein was detected in WAT, and fructose-diet-fed rats displayed higher p62 levels than the standard-diet-fed rats (three-way ANOVA; p < 0.001; Fig. 9).

The plasma leptin level and leptin mRNA expression in WAT. The whiskers indicate the range from minimum to maximum values. Both the plasma level (p = 0.005) and mRNA expression (p = 0.020) of leptin were decreased by IUGR. The leptin mRNA level was decreased by fructose consumption (p < 0.001). The abbreviations are explained in Fig. 1, n = 12–14/group

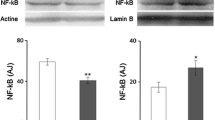
LEPR mRNA expression in a WAT and b liver. The primers used here amplify all the isoforms of LEPR. The whiskers indicate the range from minimum to maximum value. The expression is relative to GAPDH and 18sRNA expression. LEPR expression was downregulated in WAT (p < 0.001) and upregulated in liver (p < 0.001) in fructose-fed rats. IUGR resulted in upregulation of LEPR in WAT (p < 0.001). The abbreviations are explained in Fig. 1, n = 12–14/group

Expression of autophagy genes mRNA. a ATG7 expression in WAT was downregulated by the fructose diet (p < 0.001) and upregulated by IUGR (p = 0.020). b ATG7 expression in liver was increased by the fructose diet (p = 0.024) and decreased by IUGR (p = 0.007). c LAMP2 expression in WAT was slightly downregulated in fructose-fed rats (p = 0.004) and also by IUGR (p = 0.031). d LAMP2 was upregulated in liver by the fructose diet (p < 0.001). In WAT, the expression is relative to GAPDH and 18sRNA expression and in liver relative to HPRT and UBC expression. The whiskers indicate the range from minimum to maximum values. The abbreviations are explained in Fig. 1, n = 12–14/group

LC3 Western blot results. a In WAT, only LC3-II band was seen, and b in liver, both LC3-I (upper band) and LC3-II (lower band) were visible. c LC3-II intensities in WAT and d LC3-II/LC3-I ratio in liver did not differ statistically between the groups. The whiskers indicate the range from minimum to maximum values. The abbreviations for groups are explained in Fig. 1, n = 6–7/group and plus sign (+) indicates the positive control of LC3. Figure also shows β-actin staining for the blots, but as β-actin seems to be affected by fructose diet, the results are analyzed relative to total protein

p62 Western blot results in WAT. a p62 band was seen in WAT, and b the protein level of p62 was significantly higher in fructose-fed animals suggesting reduced amount of autophagy (p < 0.001). The whiskers indicate the range from minimum to maximum values. The abbreviations for groups are explained in Fig. 1, n = 6/group and plus sign (+) indicates the positive control sample. Figure also shows β-actin for the blot but as β-actin seems to be affected by fructose diet, the results are analyzed relative to total protein
In the liver, fructose consumption induced the general LEPR expression (three-way ANOVA; p < 0.001), as shown in Fig. 6b, which was seen also in the increase in LEPR-b and LEPR-f expression (three-way ANOVA; p = 0.001 and p < 0.001, respectively). Fructose also increased liver ACC, ATG7, MAP1LC3β and LAMP2 mRNA expression (three-way ANOVA; p = 0.010, p = 0.024, p = 0.003 and p < 0.001, respectively; Fig. 7b–d). Both LC3-I and LC3-II bands were visible in the Western blot of liver (Fig. 8b), but no differences caused by fructose consumption were detected in the protein levels of LC3-II or in the ratio of LC3-II/LC3-I (three-way ANOVA; p = 0.597 and p = 0.305; Fig. 8d). In the liver, p62 protein could not be detected.
Effects of intra-uterine growth restriction
Plasma leptin levels were affected by IUGR (three-way ANOVA; p = 0.005). This was seen in standard-diet-fed rats as IUGR induced a reduction in the leptin levels. Leptin mRNA expression in WAT was also decreased by IUGR (three-way ANOVA; p = 0.020). The plasma leptin and adipose tissue mRNA levels are shown in Fig. 5. On the other hand, LEPR-a and LEPR-f mRNA levels were both increased by IUGR in WAT (three-way ANOVA; p = 0.015 and p = 0.021, respectively) which was also seen by measuring general WAT LEPR expression that revealed a significant upregulation (three-way ANOVA; p < 0.001) especially in standard-diet-fed rats (Fig. 6a). However, the gene expression of SOCS3 in WAT was not changed by IUGR. Autophagy gene ATG7 exhibited a slight increase in mRNA expression levels (three-way ANOVA; p = 0.020) in contrast to LAMP2 that was somewhat reduced (three-way ANOVA; p = 0.031) in IUGR rats WAT (Fig. 7a–c).
Intra-uterine growth restriction did not change the general LEPR mRNA expression in the liver (Fig. 6b), although the expression of the LEPR-b isoform was enhanced in the livers of IUGR rats (three-way ANOVA; p < 0.001). However, no change in SOCS3 expression was detected. ATG7 expression was decreased by IUGR in the liver (three-way ANOVA; p = 0.007; Fig. 7b).
The connection between plasma triglycerides, leptin receptors and autophagy
Adipose tissue LEPR expression correlated with the mRNA expression of SOCS3 (r = 0.790, p < 0.001), ATG7 (r = 0.765, p < 0.001) and MAP1LC3β (r = 0.599, p < 0.001). Also in the liver, LEPR expression correlated with ATG7 (r = 0.505, p < 0.001), LAMP2 (r = 0.515, p < 0.001), MAP1LC3β (r = 0.501, p < 0.001) and ACC (r = 0.462, p < 0.001) expression.
Both ATG7 and LEPR expression of WAT and liver correlated with plasma TG levels. Plasma TG displayed a negative correlation especially with general LEPR expression (r = −0.557, p < 0.001), SOCS3 (r = −0.612, p < 0.001), ATG7 (r = −0.564, p < 0.001) and MAP1LC3β (r = −0.617, p < 0.001) in WAT and a less strong correlation with the leptin mRNA (r = −0.380, p = 0.001), LEPR-b (r = −0.289, p = 0.013) and LEPR-f (r = −0.317, p = 0.006). The TG concentrations were also positively correlated with liver gene expression. A strong correlation was seen between TGs and general LEPR expression (r = 0.706, p < 0.001), LEPR-f (r = 0.591, p < 0.001), LAMP2 (r = 0.591, p < 0.001) and MAP1LC3β (r = 0.468, p < 0.001) in the liver and weaker correlation between TGs and LEPR-b (r = 0.286, p = 0.013), ATG7 (r = 0.353, p = 0.002) and ACC (r = 0.282, p = 0.017).
The stepwise modeling with linear regression revealed that general LEPR expression in the liver and MAP1LC3β expression in WAT are the most important determinants of plasma TGs in these male rats. The modeling shows that the liver LEPR expression alone explained 49.1 % of the variation in the plasma TG levels (B = 0.863, SE = 0.102, R 2adj. = 0.491, p < 0.001). If MAP1LC3β expression in WAT was also included, then the model was even better explaining 57.5 % of the variation (B LEPRliver = 0.646, SELEPRliver = 0.109 and B MAP1LC3 WAT = −0.222, SEMAP1LC3 WAT = 0.057; R 2adj. = 0.575, p < 0.001). Weight, plasma leptin, ATG7, LAMP2 and ACC mRNA expression as well as LC3 protein in WAT and liver were also considered but did not explain the TGs. The correlations between TGs and the variables in the model are presented in Fig. 10.

The relationship between LEPR liver expression, MAP1LC3β WAT expression and the plasma triglyceride level. LEPR mRNA expression in the liver explains 49.1 % of the plasma TG levels (p < 0.001). LEPR mRNA expression in the liver and MAP1LC3β mRNA expression in the WAT together explain 57.5 % of the TGs (p < 0.001). The black squares represent standard-diet-fed rats (CCst, RCst and RRst), and the gray ones are fructose-fed rats (CCfru, RCfru and RRfru)
Discussion
Our aim was to study the underlying mechanisms of long-term fructose consumption and IUGR-induced metabolic changes. Since leptin acts in a paracrine way in adipose tissue controlling the fat accumulation, we studied the size of the adipocytes and the expression of the most abundant leptin receptor isoforms as well as the expression of SOCS3 which is an inhibitor of leptin signaling. Autophagy has also been suggested to affect lipid accumulation in adipose tissue and liver, and therefore, we also included the autophagy genes ATG7, LAMP2 and MAP1LC3β into our study.
The maximal adipocyte size correlated with the body weight as expected suggesting that our method for size analysis is reliable. However, the size of adipocytes in visceral fat displayed no difference attributable to fetal growth restriction or consumption of a fructose diet. In the case of IUGR, our result is supported by the parallel results of the previous study in which the 70 % fetally food-restricted animals had the same size of adipocytes as their controls (Lukaszewski et al. 2011). Also as we reported earlier (Malo et al. 2013) in the same study population as examined here, there was no difference in the lipid content of intraperitoneal, retroperitoneal and subcutaneous fat between the IUGR and control groups as measured by magnetic resonance imaging. However, the fructose-diet-fed rats displayed a slightly higher lipid content in the intraperitoneal and retroperitoneal adipose tissue than the controls (Malo et al. 2013), although the adipocytes of visceral adipose tissue were microscopically the same size. This might suggest that lipid accumulation occurred in the macrophages of adipose tissue or in perivascular tissue.
Fructose diet had no effect on plasma leptin levels, but leptin mRNA levels in adipose tissue were decreased. In our previous study, we observed that fructose consumption led to increased TGs and fasting insulin as well as decreased LDL cholesterol in these same animals (Malo et al. 2013). Our results are much in line with a previous study from (Shapiro et al. 2008) since they found no difference in the weight, adiposity or plasma leptin levels between the fructose-fed rats and controls, but the TG levels were elevated by fructose consumption. In another study, adipocyte-specific reduction in LEPR in mice resulted in similar features in phenotype as in our fructose-fed rats including elevated serum levels of insulin and TGs (Huan et al. 2003). In this particular study, the mice also displayed a higher body weight and larger adipocyte size as well as increased leptin expression which was not seen in our study.
In adipose tissue, the post-weaning fructose diet resulted in decreased expression of LEPR-b and LEPR-f. Also SOCS3 mRNA expression was decreased in fructose-fed rats. Since the SOCS3 expression is stimulated by leptin signaling, the decreased SOCS3 mRNA expression probably reflects the reduced LEPR-b mRNA expression. This suggests that a long-term fructose diet could alter the paracrine signaling of leptin. A fructose diet has also been shown to lead to impaired leptin signaling in the hypothalamus and at least to central leptin resistance (Shapiro et al. 2008). The leptin signaling in hypothalamus was lower, although no differences in the expression of leptin receptor were seen and it was hypothesized that this could be a result of TGs blocking the blood–brain barrier (Shapiro et al. 2008).
The physiological effect of LEPR-f isoform is still unknown. However, here, we observed LEPR-f to be the most abundant isoform expressed in WAT and also in the liver which suggests that it possesses an important function. Interestingly, the expression of LEPR-f in adipose tissue correlated negatively with the plasma TG level, and a positive correlation was detected between LEPR-f liver expression and TGs. This might indicate a central role for the LEPR-f isoform in metabolic processes. Previously, it has been shown that the adipocyte-specific reduction in LEPRs can lead to the elevated TG levels, insulin resistance and increased adiposity (Huan et al. 2003). Strong correlations between TGs, LEPRs and autophagy genes suggest that LEPR expression might play a role in regulating the plasma TG. The liver and WAT leptin system may well contribute to the plasma TG levels in an opposite manner. Liver expression of LEPR expression alone explained 49.1 % of the variation in the TG levels in this study. These results suggests that the effect of fructose consumption on TGs might be mediated via altered leptin signaling in liver and WAT.
In fructose-fed animals, the expression of autophagy genes ATG7, LAMP2 and MAP1LC3β were similarly regulated in the adipose tissue and liver as LEPR expression. Autophagy has been proposed to play a role in lipid storing to adipose tissue (Singh et al. 2009b; Zhang et al. 2009), and as the adipocyte hypertrophy would have been expected in fructose-fed animals, the downregulation of autophagy genes might explain the unchanged adipocyte size. In both adipose tissue and liver, the autophagy genes were expressed similarly as LEPR possibly pointing to a regulatory role of leptin to autophagy. A correlation between LEPR and autophagy gene expression in both adipose tissue and liver could also suggest a role for leptin in autophagy regulation. These results are supported by a previous study (Malik et al. 2011) which demonstrates convincingly that leptin regulated autophagy both in cell culture and in mice. It was postulated that leptin may induce autophagy via AMP-activated protein kinase (AMPK) and inhibit it through mammalian target of rapamycin (mTOR) depending on the cell type (Malik et al. 2011).
Intra-uterine growth restriction led to a somewhat lower plasma leptin, at least in standard-diet-fed offspring, and leptin mRNA levels were decreased. LEPR-a mRNA expression was increased in the fetally undernourished groups. In a previous study, 70 % fetal food restriction was shown to decrease the expression of LEPR-b in adipose tissue (Lukaszewski et al. 2011). This was not seen in our study. Since SOCS3 expression was also unchanged, it seems that IUGR has no major effects on the paracrine functioning of leptin. In addition, the minor changes seen in autophagy gene expression by IUGR are not very clear.
The changes in the gene expression caused by IUGR needs to be evaluated cautiously as the number of the individuals in these groups might be insufficient. In addition, since the suckling period examination is lacking a group in which the offspring from control dams would have been lactated by dams that were food-restricted during pregnancy, the effect of suckling period was included in the analysis as a confounding variable. Thus, the comparison is statistically challenging. However, the investigation of the effects of fructose included a total of 74 rats (38 rats on a standard and 36 on a fructose diet) which is considered to be sufficient for detecting the changes in a reliable manner.
One must also bear in mind when interpreting these results that the level of gene expression does not necessary represent the protein levels. Measuring autophagy is also challenging. The autophagy genes showed marked expression changes in WAT and liver suggesting altered autophagy, and this was supported by p62 protein levels in the WAT when the intensity of p62 band was considered relative to total protein. This might be the most appropriate way of analyzing the results as it seems that both β-actin and GAPDH protein levels are also affected by the experimental design and might, thus, not be suitable for reference in Western blot. p62 is a substrate for autophagy and is often used to measure the amount of autophagy (Bjorkoy et al. 2009). Our results suggested that the amount of p62 protein is significantly higher in fructose-fed rats indicating reduction in autophagy and supporting the gene expression results. However, LC3-II protein levels displayed no differences between the standard-diet-fed and fructose-diet-fed rats. This might be caused by the post-transcriptional regulation. On the other hand, LC3-II is often used as a measure of autophagosome formation, but it is also degraded by autophagy, making the interpretation of the results difficult (Mizushima and Yoshimori 2007). In the liver, p62 protein could not be detected in the Western blot. This is most likely explained by the experimental design as the offspring were fasted 12 h before killing and sample collecting. Starvation leads to activation of autophagy in the liver and results in degradation of p62.
Altogether, these results indicate that as a result of the long-term fructose diet, the capacity of adipose tissue to store lipids could be compromised which is also supported by decreased gene expression of ACC in WAT, and lipids might accumulate as ectopic fat and be directed to the non-adipose tissue which should be further studied. Previously, it has been shown that tissue-specific ACC knockout is associated with reduced lipid accumulation in adipose tissue and liver (Mao et al. 2006, 2009). Lipid accumulation in the organs, on the other hand, might lead to increased insulin resistance in general, including changes in the leptin system and autophagy. The effect of fructose consumption might be partly explained by decreased adipose tissue function, and an overactivated leptin system in the liver resulting in increased liver fat and elevated TG plasma levels. Furthermore, the expression of ACC mRNA was increased in liver which might also explain the higher liver lipid content. The high levels of very low-density lipoproteins produced in the liver increasing the plasma TG levels are seen in insulin resistance (Sparks et al. 2012) which was also reported previously in our rats (Malo et al. 2013). It has been claimed that liver fat accumulation is an independent determinant of insulin resistance, also independent of weight (Seppala-Lindroos et al. 2002), and as our results suggest the fat content in the liver is indeed higher in fructose-fed rats. Insulin has also been proposed to suppress liver autophagy (Ezaki et al. 2011) and overactivation of autophagy in the liver might be evoked by the liver insulin resistance. Extensive liver autophagy results in non-selective catabolization of cytoplasmic components into amino acids and free fatty acids, which are used in gluconeogenesis and beta-oxidation (Komatsu 2012). Usually, autophagy is considered as a protective mechanism, but hyperactive autophagy has also been reported in pathological conditions such as liver inflammation in a rat model of diabetes mellitus (Hagiwara et al. 2010) and as a possible cause of hepatocyte death in anorexia nervosa patients (Rautou et al. 2008) suggesting that both diminished or excessive autophagy could be deleterious. It has also been reported that leptin treatment might have unfavorable consequences via an overactivated leptin system and an SOCS3 inhibitory effect might lead to insulin resistance, hepatic steatosis and liver fibrosis (Polyzos et al. 2011).
In conclusion, a long-term fructose diet disturbs the leptin system by decreasing the expression of LEPRs and autophagy genes in white adipose tissue and enhancing their expression in the liver of male rats. We propose that the LEPR and autophagy gene expression changes in adipose tissue and liver as well as the changes in autophagy activity contribute to the metabolic alterations including elevated plasma TG and insulin resistance seen in fructose-fed animals.
References
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS (1999) The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem 274:30059–30065
Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T (2009) Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452:181–197
Briana DD, Malamitsi-Puchner A (2009) Intrauterine growth restriction and adult disease: the role of adipocytokines. Eur J Endocrinol 160:337–347
da Silveira Cavalcante F, Gombar FM, Ferreira RV, da Silva Faria T, Costa WS, Sampaio FJ, da Fonte Ramos C (2009) Maternal protein-energy and energy-restricted diets during lactation possibly could program folliculogenesis and the ovarian expression of leptin and its different isoform receptors in rats. Fertil Steril 92:1755–1757
Dekker MJ, Su Q, Baker C, Rutledge AC, Adeli K (2010) Fructose: a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am J Physiol Endocrinol Metab 299:E685–E694
Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, Iwata J, Tanida I, Furuya N, Zheng DM, Tada N, Tanaka K, Kominami E, Ueno T (2011) Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 7:727–736
Hagiwara S, Iwasaka H, Koga H, Hasegawa A, Kudo K, Kusaka J, Oyama Y, Noguchi T (2010) Stimulation of autophagy in the liver by lipopolysaccharide-induced systemic inflammation in a rat model of diabetes mellitus. Biomed Res 31:263–271
Hietaniemi M, Malo E, Jokela M, Santaniemi M, Ukkola O, Kesaniemi YA (2009) The effect of energy restriction during pregnancy on obesity-related peptide hormones in rat offspring. Peptides 30:705–709
Huan JN, Li J, Han Y, Chen K, Wu N, Zhao AZ (2003) Adipocyte-selective reduction of the leptin receptors induced by antisense RNA leads to increased adiposity, dyslipidemia, and insulin resistance. J Biol Chem 278:45638–45650
Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, Stienstra R (2012) Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 153:5866–5874
Jaquet D, Deghmoun S, Chevenne D, Collin D, Czernichow P, Levy-Marchal C (2005) Dynamic change in adiposity from fetal to postnatal life is involved in the metabolic syndrome associated with reduced fetal growth. Diabetologia 48:849–855
Komatsu M (2012) Liver autophagy: physiology and pathology. J Biochem 152:5–15
Lukaszewski MA, Mayeur S, Fajardy I, Delahaye F, Dutriez-Casteloot I, Montel V, Dickes-Coopman A, Laborie C, Lesage J, Vieau D, Breton C (2011) Maternal prenatal undernutrition programs adipose tissue gene expression in adult male rat offspring under high-fat diet. Am J Physiol Endocrinol Metab 301:E548–E559
Malik SA, Marino G, BenYounes A, Shen S, Harper F, Maiuri MC, Kroemer G (2011) Neuroendocrine regulation of autophagy by leptin. Cell Cycle 10:2917–2923
Malo E, Saukko M, Santaniemi M, Hietaniemi M, Lammentausta E, Blanco Sequeiros R, Ukkola O, Kesaniemi YA (2013) Plasma lipid levels and body weight altered by intrauterine growth restriction and postnatal fructose diet in adult rats. Pediatr Res 73:155–162
Mao J, DeMayo FJ, Li H, Abu-Elheiga L, Gu Z, Shaikenov TE, Kordari P, Chirala SS, Heird WC, Wakil SJ (2006) Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc Natl Acad Sci USA 103:8552–8557
Mao J, Yang T, Gu Z, Heird WC, Finegold MJ, Lee B, Wakil SJ (2009) aP2-Cre-mediated inactivation of acetyl-CoA carboxylase 1 causes growth retardation and reduced lipid accumulation in adipose tissues. Proc Natl Acad Sci USA 106:17576–17581
Martinez-Beamonte R, Navarro MA, Larraga A, Strunk M, Barranquero C, Acin S, Guzman MA, Inigo P, Osada J (2011) Selection of reference genes for gene expression studies in rats. J Biotechnol 151:325–334
McMillen IC, Robinson JS (2005) Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85:571–633
Mizushima N, Yoshimori T (2007) How to interpret LC3 immunoblotting. Autophagy 3:542–545
Polyzos SA, Kountouras J, Zavos C, Deretzi G (2011) The potential adverse role of leptin resistance in nonalcoholic fatty liver disease: a hypothesis based on critical review of the literature. J Clin Gastroenterol 45:50–54
Rautou PE, Cazals-Hatem D, Moreau R, Francoz C, Feldmann G, Lebrec D, Ogier-Denis E, Bedossa P, Valla D, Durand F (2008) Acute liver cell damage in patients with anorexia nervosa: a possible role of starvation-induced hepatocyte autophagy. Gastroenterology 135:840–848, 848.e1-e3
Sengupta A, Molkentin JD, Yutzey KE (2009) FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem 284:28319–28331
Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, Goto T, Westerbacka J, Sovijarvi A, Halavaara J, Yki-Jarvinen H (2002) Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab 87:3023–3028
Shapiro A, Mu W, Roncal C, Cheng KY, Johnson RJ, Scarpace PJ (2008) Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am J Physiol Regul Integr Comp Physiol 295:R1370–R1375
Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E, Uchiyama Y (2009) The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 382:419–423
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009a) Autophagy regulates lipid metabolism. Nature 458:1131–1135
Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ (2009b) Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 119:3329–3339
Sparks JD, Sparks CE, Adeli K (2012) Selective hepatic insulin resistance, VLDL overproduction, and hypertriglyceridemia. Arterioscler Thromb Vasc Biol 32:2104–2112
Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI (1995) Identification and expression cloning of a leptin receptor, OB-R. Cell 83:1263–1271
Wang MY, Grayburn P, Chen S, Ravazzola M, Orci L, Unger RH (2008) Adipogenic capacity and the susceptibility to type 2 diabetes and metabolic syndrome. Proc Natl Acad Sci USA 105:6139–6144
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S (2009) Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA 106:19860–19865
Acknowledgments
We would like to thank Saija Kortetjärvi, Heidi Häikiö, Erja Tomperi and Mirja Vahera for expert technical assistance. This study was supported by the Finnish Foundation for Cardiovascular Research.
Conflict of interest
They have no conflict of interest.
Ethical standard
All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Äijälä, M., Malo, E., Ukkola, O. et al. Long-term fructose feeding changes the expression of leptin receptors and autophagy genes in the adipose tissue and liver of male rats: a possible link to elevated triglycerides. Genes Nutr 8, 623–635 (2013). https://doi.org/10.1007/s12263-013-0357-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12263-013-0357-3