
Abstract
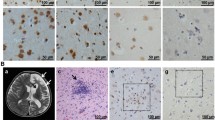
Rasmussen’s encephalitis (RE) is a rare and severe progressive epileptic syndrome with unknown etiology. Infection by viruses, including human cytomegalovirus (HCMV), has been speculated to be a potential trigger for RE. However, no viral antigens have been detected in the brains of patients with RE; thus, a possible clinical linkage between viral infections and RE has not been firmly established. In this study, we evaluated the expression of HCMV pp65 antigen in brain sections from 26 patients with RE and 20 non-RE patients by immunohistochemistry and in situ hybridization, and assessed the associations between HCMV infection and clinical parameters. Elevated expression of HCMV pp65 protein and DNA was observed in 88.5% (23/26) and 69.2% (18/26) of RE cases, respectively. In the non-RE group, HCMV pp65 antigen was detected only in two cases (10%), both of which were negative for DNA staining. Additionally, the intensity of HCMV pp65 staining was correlated with a shorter duration of the prodromal stage, younger age of seizure onset, and more severe unilateral cortical atrophy. Elevated expression of HCMV pp65 was observed in RE brain tissue and was correlated with the clinical features of RE disease. In summary, our results suggested that HCMV infection may be involved in the occurrence and progression of RE disease. Thus, further studies are needed to determine whether early treatment with anti-HCMV antibodies could modulate the course of RE.

Article PDF
Similar content being viewed by others


Avoid common mistakes on your manuscript.
References
Atkins MR, Terrell W, Hulette CM. 1995. Rasmussen’s syndrome: a study of potential viral etiology. Clin Neuropathol, 14: 7–12.
Bauer J, Bien CG, Lassmann H. 2002. Rasmussen’s encephalitis: a role for autoimmune cytotoxic T lymphocytes. Curr Opin Neurol, 15: 197–200.
Bien CG, Bauer J, Deckwerth TL, Wiendl H, Deckert M, Wiestler OD, Schramm J, Elger CE, Lassmann H. 2002a. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen’s encephalitis. Ann Neurol, 51: 311–318.
Bien CG, Granata T, Antozzi C, Cross JH, Dulac O, Kurthen M, Lassmann H, Mantegazza R, Villemure JG, Spreafico R, Elger CE. 2005. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain, 128: 454–471.
Bien CG, Widman G, Urbach H, Sassen R, Kuczaty S, Wiestler OD, Schramm J, Elger CE. 2002b. The natural history of Rasmussen’s encephalitis. Brain, 125: 1751–1759.
Britt W. 2015. Controversies in the natural history of congenital human cytomegalovirus infection: the paradox of infection and disease in offspring of women with immunity prior to pregnancy. Med Microbiol Immunol, 204: 263–271.
Chen S, Chen S, Guan Y, Zhang Y, Qi X, An J, Wang Y, Luan G. 2016. Elevated expression of human papillomavirus antigen in brain tissue of patients with Rasmussen’s encephalitis. Epilepsy Res, 126: 119–125.
Farrell MA, Cheng L, Cornford ME, Grody WW, Vinters HV. 1991. Cytomegalovirus and Rasmussen’s encephalitis. Lancet, 337: 1551–1552.
Gahring L, Carlson NG, Meyer EL, Rogers SW. 2001. Granzyme B proteolysis of a neuronal glutamate receptor generates an autoantigen and is modulated by glycosylation. J Immunol, 166: 1433–1438.
Guan Y, Zhou J, Luan G, Liu X. 2014. Surgical treatment of patients with Rasmussen encephalitis. Stereotact Funct Neurosurg, 92: 86–93.
Jay V, Becker LE, Otsubo H, Cortez M, Hwang P, Hoffman HJ, Zielenska M. 1995. Chronic encephalitis and epilepsy (Rasmussen’s encephalitis): detection of cytomegalovirus and herpes simplex virus 1 by the polymerase chain reaction and in situ hybridization. Neurology, 45: 108–117.
Li Y, Uccelli A, Laxer KD, Jeong MC, Vinters HV, Tourtellotte WW, Hauser SL, Oksenberg JR. 1997. Local-clonal expansion of infiltrating T lymphocytes in chronic encephalitis of Rasmussen. J Immunol, 158: 1428–1437.
Looker KJ, Magaret AS, Turner KM, Vickerman P, Gottlieb SL, Newman LM. 2015. Global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS One, 10: e114989.
Luan G, Gao Q, Guan Y, Zhai F, Zhou J, Liu C, Chen Y, Yao K, Qi X, Li T. 2013. Upregulation of adenosine kinase in Rasmussen encephalitis. J Neuropathol Exp Neurol, 72: 1000–1008.
Luan G, Gao Q, Zhai F, Chen Y, Li T. 2016. Upregulation of HMGB1, toll-like receptor and RAGE in human Rasmussen’s encephalitis. Epilepsy Res, 123: 36–49.
Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The "silent" global burden of congenital cytomegalovirus. Clin Microbiol Rev, 26: 86–102.
Merkler D, Horvath E, Bruck W, Zinkernagel RM, Del la Torre JC, Pinschewer DD. 2006. "Viral deja vu" elicits organ-specific immune disease independent of reactivity to self. J Clin Invest, 116: 1254–1263.
Pardo CA, Vining EP, Guo L, Skolasky RL, Carson BS, Freeman JM. 2004. The pathology of Rasmussen syndrome: stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia, 45: 516–526.
Power C, Poland SD, Blume WT, Girvin JP, Rice GP. 1990. Cytomegalovirus and Rasmussen’s encephalitis. Lancet, 336: 1282–1284.
Rasmussen T, Olszewski J, Lloydsmith D. 1958. Focal seizures due to chronic localized encephalitis. Neurology, 8: 435–445.
Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, Hughes TE, Heinemann SF, McNamara JO. 1994. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science, 265: 648–651.
Schwab N, Bien CG, Waschbisch A, Becker A, Vince GH, Dornmair K, Wiendl H. 2009. CD8+ T-cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain, 132: 1236–1246.
Takahashi Y, Matsuda K, Kubota Y, Shimomura J, Yamasaki E, Kudo T, Fukushima K, Osaka H, Akasaka N, Imamura A, Yamada S, Kondo N, Fujiwara T. 2006. Vaccination and infection as causative factors in Japanese patients with Rasmussen syndrome: molecular mimicry and HLA class I. Clin Dev Immunol, 13: 381–387.
Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol, 20: 392–401.
Varadkar S, Bien CG, Kruse CA, Jensen FE, Bauer J, Pardo CA, Vincent A, Mathern GW, Cross JH. 2014. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol, 13: 195–205.
Walter GF, Renella RR. 1989. Epstein-Barr virus in brain and Rasmussen’s encephalitis. Lancet, 1: 279–280.
Acknowledgments
This work was supported by the following funds: the National Natural Science Foundation of China (81571275), the Beijing Municipal Natural Science Foundation (7144217), the Capital Applied Clinic Research Programs of Science and Technology (Z131107002213171), the Beijing Rising-star Plan of Science and Technology (Z141107001814042), the Open Research Fund of the Beijing Key Laboratory of Epilepsy Research (No. 2014DXBL02), Capital Medical University (15JL08), Scientific Research Common Program of Beijing Municipal Commission of Education (KM201610025001), Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (2014 1685).
Author information
Authors and Affiliations
Corresponding authors
Additional information
These authors contributed equally to this work.
ORCID: 0000-0002-9674-6549
ORCID: 0000-0002-2946-7371
This article is published with open access at Springerlink.com
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, Y., Wang, Y., Chen, S. et al. Expression of human cytomegalovirus components in the brain tissues of patients with Rasmussen’s encephalitis. Virol. Sin. 32, 115–121 (2017). https://doi.org/10.1007/s12250-016-3917-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12250-016-3917-z