
Abstract
The advent of the twenty-first century marked a paradigm shift in the healthcare sector with coming of automated, sensitive, targeted medicines and technologies having diagnostic, prophylactic and therapeutic effects. Nanomedicines also attained wide acclamation in their initial years, but the transformation from being the proof of concept to successfully marketed products seems very daunting. Although the reason for this may be attributed to slow but incremental character of many present-day technologies, the review asserts that there are other significant facets that may purvey a thorough explanation of this scenario. The article elaborately discusses the hurdles hindering clinical translation of nanomedicines including scale-up challenges, in vitro in vivo cascade of toxicology assays, along with unrefined manufacturing guidelines, inadequate regulatory approvals, competitive conventional market, etc., leading to hesitant investments by pharmaceutical giants. The paper also explores the economic viability of nanobiotechnology sector through an empirical investigation of the revenue data of various pharmaceutical industries manufacturing nano-based drugs, which indicates minor commercial importance of these medicines. We also laid down a comprehensive set of recommendations to smoothen the translational pathway of nanomedicines from an idea to reality, efface the consumer distrust and push boundaries for development and launching of safe, efficient and commercially successful products.

Graphical abstract
Similar content being viewed by others


For the past few decades, nanobiotechnology has been proclaimed as the ‘revolution in making’ with the potential impact being compared with groundbreaking innovations like penicillin, smallpox and polio vaccine or injectable insulin. Owing to potential promises of highly accurate, targeted, optimized and personalized treatment, nanopharmaceuticals created a buzz in the market. The coming of FDA-approved anticancerous drug Doxil in 1995 proclaimed the dawn of nanomedicines [1]. The inclusion of drug delivery vehicles and medico-diagnostic devices with immense therapeutic, prophylactic and diagnostic applications at nanoscale attracted the attention of not only the scientific community but also the pharmaceutical sector. Today, there is a vast variety of nanoparticles employed in the field of medicine that can be broadly classified into two broad categories: organic and inorganic nanoparticles. The organic nanoparticles can be further divided into polymeric, liposomal, micelles, dendrimers, magnetic nanoparticles, protein-based nanoparticles, theranostic nanoparticles, mesoporous silica nanoparticles, carbon nanotubes, etc. Further, inorganic nanoparticles can be categorized as quantum dots and metallic nanoparticles like silver, gold, iron oxide, titanium, etc. [2, 3]. The share of different categories of FDA-approved nanomedicines until 2016 is represented in Fig. 1. The higher efficacy of drugs with minimal dosage along with targeted drug delivery formed the base of all recent nanomedicines [4]. These nanomedicines have shown potential in the treatment of many life-threatening diseases such as cancer, Alzheimer’s, Parkinson disease, hepatitis B, hepatitis C, rheumatoid arthritis, SCID, multiple sclerosis, psoriatic arthritis, chronic gout, Karposi’s sarcoma, haemophilia, acute lymphoblastic leukaemiaand psychostimulant [5,6,7,8,9].

Categories of successfully marketed nanomedicines. The pie charts represent the proportion of nanomedicines approved by FDA in different categories. A total of nearly about 51 nanopharmaceuticals approved till 2016 were considered. The legend gives abbreviations for the categories of nanomedicines
Nanobiotechnology-based products constitute a fast-growing market, which encompasses a combination of benefits, risks, promises and uncertainties [10]. The global nanomedicines’ market is expected to grow at a CAGR of 17.1% in the forecast period spanning from 2017 to 2023. Their empowering capacity in the healthcare sector is expected to create USD 392.8 billion by 2023 [11]. But the important question is, has the technology really lived up to our expectations? The recent inflow of reports on nanobiotechnology and nanomedicines, especially related to the pharmaceutical sector, has created the impression of its immense economic viability. Though a substantial contribution of nanopharmaceuticals to cardiology and oncology can be ascertained through significant clinical success, the drastic impact is still awaited [12]. The need of the hour is to check to what extent this new technological revolution has contributed to the intellectual property portfolio of the big market giants. The capitalizing abilities of nanobiotechnology need to be critically analysed to ascertain the hype associated with it. Despite trying different permutations and combinations of nanoparticles, that it may uncover breakthrough properties, the palpable shift in diagnostics and treatment of diseases is yet to be seen. Moreover, the innovations pertaining to this sector are always met with a speed bump, called toxicological analysis. However, little is known about the safety data, toxicity profiles, health risks and their plausible interaction with the biological systems and environment [13]. It is important here to check how big is this issue which concerns animals, humans and environmental health on the whole. This has led to more exhaustive, elaborate and costly testing of nanomedicines to establish the health risks pertaining to its use. The approval of nanomedicines also faces regulatory constraints due to the absence of set forth data requirements, tests and practical assays by the FDA and other regulatory agencies [14]. The coming of FDA’s non-binding Draft Guidance Document for drug products containing nanomaterials in 2017 finally laid out efficacy and safety parameters to be met, but the infancy of this field is clearly reflected with persistent wariness felt by pharmaceutical giants while deciding approval pathways of these medicines [15, 16].
The Reality Check of Nanomedicines: Benefit to Risk Assessment
The use of nanomedicines either directly as therapeutic agents or as drug delivery agents (nanocarriers), active implants, contrasting agents for bioimaging and diagnostics, drug encapsulates, gene delivery systems and tissue engineering has brought immense impetus to pharmaceutical and medical research [17]. In the last decade, the scientific community expressed a high commitment to exploring the medical application of nanotechnolgy through an exponential rise in the number of research articles and review papers [18]. Although more than 280 products of medical applications containing nanomaterials have been approved by FDA, with around 100 nanomedicines already sold in the market, yet the meaningful performance of nanomedicines in the market needs to assessed [19].
The market for nanomedicines initially progressed due to a few major limitations of conventional drug delivery system. Off-site drug release, strenuous removal of the residual non-biodegradable material that is left within the patient’s body causing toxicity, unsustainable burst of drug release instantaneously after administration and low solubility are few of the restricting factors of conventional drugs. One single solution to all the above-mentioned problems was found in nanomedicines, which are either consumed directly using the nanocrystals as a therapeutic agent or encapsulating the therapeutic drug with nanoparticles [20]. The broad majority of nanomedicines in preclinical and clinical development as well as in clinical applications are used for targeting a specific tissue or an organ, usually cancer or a tumour site. Nanocarriers offer many distinct advantages like a higher percentage of drug loading owing to large surface area, reduced overall drug dose which significantly reduces the potential side effects, higher active concentration in the living system providing an enhanced bioavailability and better dissolution of drug along with a cost-effective formulation [21]. Owing to these perks, a number of big pharmaceutical companies such as Abbvie, Jansen, Sanofi, Amgen, Merk and Celgene are putting huge investment in this sector. These advantages are often countered with numerous negative factors such as non-predictive pharmacology, bioavailability, cellular uptake, degradation and clearance at the target site, toxicity and unclear safety profile of nanopharmaceuticals [22, 23]. A balance needs to be achieved for advanced and progressive outcomes. Therefore, in spite of being a commercial success, neither Doxil, a PEGylated liposomal antitumour nanomedicine revolutionized the map of chemotherapy, nor did the nanocrystalline drugs like Rapamune or the nanocarrier-based therapeutics like Ambiosome or Amraxane [24].
Based upon its ability to enhance Intellectual Property portfolios through patent procurement, the developmental time and cost requirement, physicochemical stability, potential long-term and short-term toxicities, ethical challenges, environmental impact during manufacture, processing, and disposal and government regulations need to be analysed to clearly decipher the market presence of nanomedicines. Moreover, the most important factor for promotion of nanoparticles lied in its enhanced and favourable properties which significantly differed from bulk material. But the same asset can be viewed as a problem when pharmacokinetic properties of nanomaterials are not understood owing to the same reason.
In light of these uncertainties, the article critically reviews the position of nanomedicines which is though consolidated, but not expanding at the desired rate. In defiance of the benefits offered by nanomedicines over conventional drug delivery systems (CDDSs), it is firmly believed that the industrial sector wants to invest in fields where the approvals with regard to therapeutic agent and nanoparticle have been already met. There is no doubt regarding the fact that the pharmaceutical industries thrive on the shoulders of giants, that is, they leap forward into this market by making nanoformulations of the existing drugs. The claim can be validated by checking almost all the 51 nanodrugs approved by FDA till 2016. Most of these drugs were modifications of existing and marketed therapeutic agents than new nanoformulations providing novel pharmacological effect [25]. The benefit-to-risk evaluation of nanomedicines is reviewed considering the hurdles offered to translation of nanomedicines from therapeutic idea to clinical use. Moreover, the current market trends with respect to nanomedicines, considering that the available challenges which influence the overall acceptance, manufacture and sales of nanopharmaceuticals are also precisely discussed.
Factors Influencing the Market Trends of Nanomedicines
Undeterred by the significant therapeutic advantages of nanopharmaceuticals for plenty of biomedical applications, their clinical translation has not thrived as expeditiously as the multitude of positive preclinical results would have propounded. Thus, in order to enhance the worth of nanomedicines in the complete pharmaceutical sector and bring them into the mainstream market, many challenges need to be addressed.
Valley of Death in Nanomedicines: a Costly Affair
Considering the therapeutic prospect of nanomedicines, the accumulation at the target site and least toxicity to the system are two key parameters that need to met. Often, in the presence of an efficient drug, nanocarriers or encapsulation simply enhance the process without changing the therapeutic component. This involves amalgamation or fusion of different nanocarriers preexisting in the market for the required drug. The total cost for the complete manufacture of new nanopharmaceutical drug in its lifetime before it reaches the market consists of the effort, time and money for development of the original idea and preclinical research, determining the potential of industrial synthesis, crossing the ‘valley of death’ which is the period of highest expenses owing to vigorous human clinical trials, and finally attaining the regulatory approval, followed by the phase of commercialization and marketing. But similar to all pharmaceutical drugs, there is resolute effort required to cross valley of death for candidate molecules of nanoscale to reach from lab bench side to bedside. According to a recent study, the success rate of nanodrugs is only 11.2%, that is, 80–90% of the pharmaceutical drugs eventually fail even after approval of clinical trials [26, 27]. The high rates of failure clearly justify the increasing costs of developing any new pharmaceutical drug. This is especially relevant to the added complexity of novel nanopharmaceuticals which has distinct properties at nanoscale.
Another aspect to be considered here is the reluctance of big pharmaceutical giants to invest in risky ventures like nanodrugs. These companies have all the resources at their disposal in terms of efficient team of researchers and scientists, bioprocess engineers to optimize manufacturing procedures, responsible set of lawyers for timely filing and defending of patents and large funding from shareholders and profit from already marketed drugs for launching and advertisement of new drug. But it is an established fact that the root of all impending inventions is small ventures. The small start-ups usually have government funding at the beginning for research and drug development, but the technical limitations in terms of high-end technologies, costly lawsuits and inadequate resources may prohibit them from venturing into this market without the assistance of larger companies [28]. Even though the efficacy and cost-effectiveness of drugs may lead to enormous profits, however, the high failure rate has deterred the investments in this area.
Government Funding and Investments: Develop or Commercialize?
According to the President’s budget of 2020 in the USA, the amount of USD 1.4 billion has been requested for the National Nanotechnology Initiative (NNI), which is the parent organization funding National Institutes of Health (NIH) involved in nanobiotechnology-based healthcare research. From the year of its inception in 2001, NNI has cumulatively given USD 29 billion for development and application of nanosciences [29]. In Europe, the research and development of nanomedicines are widely supported by the European Commission through two agencies, namely, Framework Programmes for Research and Development (FP7) and Horizon 2020. The healthcare sector has received around Euro 400 million under 85 projects of Nanosciences, Nanotechnologies, Materials and New Production Technologies (FP7-NMP) and Euro 150 million for 31 projects under FP-7 Health [30]. The funding for nanopharmaceuticals is also provided by a number of governments around the globe. The developing countries like India which began with initiatives like 5-year programme called Nano Mission with large funding of USD 250 million are investing huge amounts in academia as well to sponsor development in this sector through National Mission on Nanosciences and Nanotechnology [31]. Australia and Japan are also pouring money for creating a market of nanomedicines. But the matter of concern is the clinical translation of these developments into products and their acceptance in the market. Regardless of the availability of resources to the research organizations, the lack of trust and communication amongst the academic and industrial sectors results in wastage of scientific efforts and lost opportunities for advancement in the pharmaceutical industry.
Investments by Pharmaceutical Giants: the Game of Uncertainty
Unlike the trending high-tech software, IT and electronic start-ups, which are capable of launching an exponential variety of new products within months, a biotechnological/nanopharmaceutical start-up often entails years of scrupulous funding to move the products from discovery to clinical trials towards regulatory approval. Tens to hundreds of millions are lost to failed clinical trials, depending upon the stage of development and trial size. The missing of primary endpoints in the clinical trials always results in plunged stocks, stock sell-offs, layoffs, incompetency in raising additional capital and, in some cases, closure and fire sale of assets in the company.
It is very difficult to accurately evaluate the investments made from capital markets, venture capital (VC) and big pharmaceutical companies in the development of nanomedicines. Even the approved and marketed nanodrugs have merged/changed hands and faced threats of being terminated multiple times on their road to market approval. For instance, Abraxane, Depocyte and Doxil brought in USD 950 million, USD 25 million and USD 252 million respectively, in 2018. But despite of creating a huge market, the history of Doxil accounts for a number of challenges. After being developed by Sequus Pharmaceuticals founded in 1981, the medicine got approved in 1995 [32]. Within a period of 3 years, ALZA Pharmaceutical acquired Sequus Pharmaceuticals in a USD 580 million deal [33], which thereafter culminated into a merger with Johnson & Johnson in 2001 for USD 10.5 billion [34]. Another such example is Accurins, a nanomedicine to treat cancer which was developed by BIND Therapeutics. It raised around USD 800 million from capital markets, venture capital and partnership with bigger pharmaceutical industries [35]. Yet, the unsatisfactory results and failure in the phase 2 trials led to the termination of partnership with Amgen [36, 37], plunging of stocks, stocks sell-off and subsequent acquisition by Pfizer for USD 40 million [38]. Two industries that are Arrowhead Pharmaceuticals and Merrimack Pharmaceuticals were shedding a whooping sum of 4.6 and 2.1 billion dollars for funding the development of nanomedicines, are yet to bear the fruits of success [39]. Arrowhead Pharmaceuticals, which was developing nanomedicines for chemotherapy and was fairly valued at USD 3 billion, recently saw a dip of 14.2% in the stock market [40] with all 12 drugs in the pipeline lying in the vicious loop of Phase trials. Another industry focused on liposomal chemotherapeutic drug delivery, Merrimack Pharmaceuticals, discontinued the development of antibody-directed liposomal drugs MM-302 in 2016 because of negative outcomes [41] and MM-310 in 2019 owing to its toxicity [42]. Moreover, the company’s product ONIVYDE, known for its treatment in pancreatic cancer, was out licensed by Ipsen in 2018 for around USD 1 billion [43]. Currently, the company has no nanoformulation-based medication under development. Thus, the technology predicted with an abundance of long-term potential on the paper needs time and effort along with caution to demonstrate the efficiency in the real world. Moreover, if the developers or innovators do not get the desired monetary benefits owing to limited resources and market standing, the advancements in the nanopharmaceutical field may be thrown at the back foot.
Biological Safety and Compatibility: a Perturbed Situation
The nanomaterials in pharmaceutical industry occupy an exceptional niche, where the diagnostics and treatment are not the only focus of medicine. The exposure of humans, animals, ecosystem and environment on the whole needs to be checked at manufacturing, processing, treatment and even disposal stage. The interface between toxicology and nanomaterials has led to the emergence of a sub-discipline, called nanotoxicology. Now, nanotoxicology comprises of three main domains; the first reflect upon the unique physicochemical properties of nanomedicines, and the second deals with in vitro and in vivo studies for toxicological assessment followed by clinical trials and finally the alternative testing strategies to check the efficiency, design and manufacture of nanomedicine/nanomedical types of equipment.
Nanomedicine Characterization
In most nanopharmaceutical drugs, the active substance is often not a homo-molecular structure. Similar to complex biological products such as monoclonal antibodies and therapeutic proteins, their structure generally consists of closely related but different entities that cannot be easily isolated, characterized, quantified and described by physicochemical analysis. The common physicochemical properties include their mean size, size distribution, shape, surface charge, surface area, chemical composition, hydrodynamic nature, aggregation tendency, solubility, surface roughness and porosity [44, 45]. These play an important role in their interactions with biological systems, but the status of current studies does not ascertain the fate of nanoparticles. The three prime combinatorial factors (characteristics of nanoparticles, environmental parameters and target cell properties) involved in toxicity assessment of nanoparticles make it difficult to determine the reliable and reproducible health risks associated with exposure of nanomedicines.
Thus, one set of rules and standards cannot be made to optimize both PEGylated and liposomal nanomedicines. Likewise, dendrimers, quantum dots, carbon nanotubes and metallic nanomedicines cannot be treated at par. The characterization of nanomedicines uses a number of optical, electronic and hydrodynamic analyses. When applying for a specific technique, several factors need to be considered such as selectivity, specificity to certain materials, precision, sensitivity, sample preparation, non-destructive nature, availability and cost.
The first issue of characterization begins with a variability of the physicochemical properties of nanomedicines in a cell-free media and biological environment. The microenvironment of biological system such as body fluid composition, immune cells, proteins, complement fragments and blood vessel structure influence the nanodrug’s carrier structure, clearance rate, drug dissociation profile and may also contribute to destabilization of the drug resulting in early, uncontrollable release of the encapsulated drug [46]. Moreover, the health status of patients, cardiac diseases, diabetes mellitus, blood pressure and genetic makeup play an important role in determining the efficiency of nanomedicines [47]. The prominent use of polyethylene glycol (PEG), as a carrier for nanomedicines, is mainly attributed to its ability in increasing the half-life and stability of the drug by improving the circulation time and evasion by the immune system. The examples of widely used FDA-approved PEG liposomes are Mitoxantrone (Novantrone), Doxil and Lipoplatin. However, there are few examples of drugs entrapped within nanocarriers, which were expected to be stable in biological fluids according to in vitro stability studies like colloidal stability, critical micelle concentrations (CMCs) and drug release profile, but show poor drug retention in circulation that is being released in less than 1 h in blood circulation. The examples for the same are albumin-based nanomedicine containing paclitaxel, abraxane, PEG-PLGA-entrapped doxorubicin, etc. Thus, the instability of these drugs not only limits therapeutic specificity, the stability of these nanocarriers but may also cause undesired/cytotoxic effects in the living system. So, the characterization assays need to be performed not only at manufacturing stage but also at diagnostic stage. The advantages and toxicological effects caused by different categories of nanomedicines are elucidated in Table 1.
In vitro and In vivo Toxicity Assessment
In vitro assays are crucial for investigating the biochemical mechanisms and effects of nanomedicines on living entities. There are numerous ways in which nanoparticles potentially damage the living cells such as cellular membrane disruption, production of reactive oxygen species, DNA damage, mitochondria malfunction, apoptosis pathway activation, genotoxicity and release of cytokines causing inflammation [48]. Not only cells but the tissue organization and organ integrity may be rendered inutile due to intrusion of nanoparticles. The ability of nanomedicines to cross the blood-brain barrier again becomes questionable when the intention is de trop.
Notwithstanding the inherent advantages like low cost, timely acquisition of results, minimal ethical concerns and efficient in vitro assays involved in the real-time detection of the mechanisms involved in the generation of free radicals, DNA lesions and degradation products, the cell cultures do not completely mimic the inherent tissue microenvironment, and thus the results of conventional cell-based assays are not completely reliable [49].
In vivo assays are generally used to determine the unintentional uptake of nanomedicines (designed to treat peripheric diseases) through the blood-brain barrier and their potential toxic effects using models like drosophila, zebrafish, rodent, canine and non-human primates [50]. The assessment uses a variety of techniques to analyse the uptake of drugs by an unintended site like brain, for instance, microdialysis, cerebrospinal fluid sampling, brain uptake index, blood/plasma ratio determination, quantitative autoradiography and imaging techniques [48]. One such example is Venofer, which was introduced in the 1950s as the first intravenously injected nanocolloidal solution of iron product. In order to reduce the high toxicity of polynuclear Fe(III)-oxyhydroxide, the core was stabilized by a carbohydrate shell. This increased the tolerance of Venofer by more than 20-fold at a 50% higher lethal dose when tested in mice [51]. But the results need credible approval in human systems owing to different microenvironments, genetic makeup and disease conditions which may influence the behavioural patterns of nanomedicines.
In spite of the therapeutic efficiency and credibility of nanomedicines established via preclinical and clinical trials, there is a long list of drugs that were discontinued after being in the market for years. For example, a protein-based nanodrug, Ontak, was approved by FDA in the year 1999 to treat leukaemia and T cell lymphoma [52]. It was used to deliver diptheria toxin selectively to target cells that expressed interleukin-2 receptors. Contrary to the benefits, the drug also caused hypersensitivity reactions, fever, nausea, vomiting, abnormalities in the blood test, poor appetite, liver problems and shortness of breath, etc. In 2006, it was found that the use of Ontak resulted in a loss of vision, and thus, its marketing was discontinued in the USA in the year 2014 [53]. Similarly, a metallic nanopharmaceutical Feridex, containing iron oxide nanoparticles, was extensively used as an MRI contrast agent. Owing to its chronic side effects, the production of Feridex was discontinued in 2008 [54]. The list also includes Feruglose® (NC100150) and Resovist®, which have been withdrawn due to their safety concerns in the long term [55].
This ascertains that there is an inadequate understanding of the relation between physicochemical properties and clinical pharmacokinetics of nanomedicines. Likewise, it also represents the insufficiency of conventional animal models to correctly predict and extrapolate the biodistribution and toxicity of nanomedicines to humans. This is especially relevant when comparing a novel nanopharmaceutical drug with conventional formulations, or when evaluating a generic version of an approved nanomedicine versus the original innovator product.
Alternative Testing Strategies
In order to address the biocompatibility and safety limitations of nanomedicines, the need of the hour is the compulsory introduction of alternative testing strategies. In vitro assays have progressed with the introduction of novel strategies in the field of tissue/organ engineering such as three-dimensional bioprinting, organs-on-a-chip and organoids [56, 57]. The toxicity analysis of engineered nanoparticles is using high throughput screening for determining morphometric, functional and biochemical properties of cells. Tests like species sensitivity distribution (SSD) to estimate the maximum acceptable concentration of nanomedicines in the environment and band gap analysis (BGA) to check the chronic effects of metallic nanomedicines are also making progressive advancements in establishing the safety of the drugs. Further, OMICS can be used to identify new pathways and mechanisms involved in nanotoxicity which could not be determined using conventional technologies. Moreover, companies need to introduce novel approaches like quality by design (QBD), quantitative structure-activity relationships (QSARs) and computational models that link specific properties or structures to the elicited responses for faster and reproducible results [58, 59]. The efforts are also being done in deciphering the potential hazards of drugs containing nanoparticles before their clinical assessment through the development of predesigned, computational and regulatory models such as NanoTEST, NANoREG and NANoREG2 projects [56]. These have been particularly undertaken to design an innovative framework in order to facilitate easy naming, categorization and classification of nanoparticle containing products along with measuring their toxicity and environmental impact. This will not only ensure the social welfare but also reduce the effort, time and financial burden incurred by pharmaceutical companies in launching their nanomedicines.
From Laboratory Bench Side to Bedside: Scale-up
The slow-paced clinical translation of nanopharmaceuticals can mainly be attributed to the three-dimensional multi-component structure of the drug itself. During the developmental stage of Investigational New Drug (IND), a small number of nanodrugs are synthesized with definite achievable reproducibility, accurate and optimized characteristics. But when manufactured at large scale, there are potential issues which affect the quality of nanodrugs which includes (1) lack of developmental guidelines, (2) inconsistent batch to batch reproducibility, (3) inadequate developmental pathways for in vitro synthesis of drugs, (4) inefficient infrastructure and in-house expertise of sterilization techniques and (5) environmental exposure during manufacture of nanomedicines.
Non-specified Manufacturing Guidelines
The essential requirement to produce large-scale nanomedicines is a proper production method that allows consistent manufacture with high-quality and batch-to-batch reproducibility. A broad array of processes are involved in the synthesis of nanomedicines like ultrasonication, high-speed homogenization, centrifugation, milling, emulsification, UV irradiation, microwave-assisted synthesis, cross linking, evaporation of organic solvents, ultrafiltration and lyophilization [60]. Though, the researchers have got a good hold over designing and controlling the manufacture, the technical advancement is yet to bear its fruits. The consolidation of the above claim can be attained from the top notch nanomedical therapeutic Doxil [61]. The medicine had to be suspended in November 2011 because of manufacturing and sterility issues. Nevertheless, the shortage of drugs was overcome with the development of alternative manufacturing approaches in 2014 but it led to delayed treatment to patients and increased medication costs [62].
The structural and physicochemical complexities of the individual components in the formulations, their specific arrangement and interaction amongst themselves offer a series of obstacles in scale-up of nanopharmaceuticals. Manufacture of useful conformations of nanomedicines with key attributes and functions requires the identification of critical process steps, key analytical criteria and important regulatory checkpoints at the initial stages itself. The process also needs to be simple with least manufacturing steps [63]. The production of the drug becomes more challenging with the inclusion of multiple targeting components, addition of surface modification with ligands and/or coatings, or by encapsulation of more than one therapeutic agent. The multistep production process not only makes the quality control and quality assurance evaluation of the products difficult, but it also poses inevitable problems for following Good Manufacturing Practices in large-scale production [64, 65].
In situ Assembled Nanoformulations
Another challenge that needs to be addressed is the reproducibility of the nanomedicine formulation when prepared in situ. The coming of magnetic, self-assembled nanomedicines gained a lot of popularity in the last decade, which were made up of two or more individual components. These intermediary components when brought together under appropriate physical and chemical conditions in the body, formed structures or complexes as the finished product suitable for addressing the disease in humans [66,67,68]. The otherwise complex manufacturing steps involved to produce stable nanopharmaceutical drug were circumvented using this technique, which significantly reduced the cost and complexities of manufacturing.
Nonetheless of these advantages, the technique fails to answer a number of queries like, can pharmaceutical companies and regulatory authorities rely on individual idiosyncrasies in the patient, doctor’s clinic or hospitals to manufacture reproducible finished products, if the complex 3D structure of nanomedicines is critical to their function? Would the final products be subjected to the same standards and stringent quality checks to ensure their functionality, therapeutic efficacy and stability? Are the pharmaceutical companies using patients to perform an important part in the manufacturing of the drug? And most importantly, should there be qualifying clinical approvals for these products as for shelf stable nanomedicines? The evolution in the scope of nanomedicines forces careful consideration in scale-up, guidelines and policies for the new products.
Sterility or Functionality: What Is at Stake?
There are number of factors to be considered before selecting the sterilization technique of nano-based products. These factors may include the ratio of drugs, polymers, targeting moieties, the type of emulsifier, organic solvent, stabilizer, cross-linker, the oil-to-water phase ratio, pressure, temperature, pH and the mixing conditions [69]. Depending upon these conditions, the products may obtain altered conformation, coagulated or denatured therapeutic, non-functional active site or a substantial amount of impurities. Thus, the therapeutic drug containing nanoparticles should have intact structural integrity and physicochemical properties which are preserved throughout the manufacturing process to the finished formulation. The size and composition of the nanoformulations offer major challenges while the selection of sterilization technique before it is administered to the body. Sterilization techniques like gamma irradiation or autoclaving offer high risk to nanomedicines, especially when one of the components is biological material [70]. In case of liposomal nanomedicines, where the particle structure is malleable or flexible, conventional sterilization filters with average particle size well below 220 nm, may be suitable [71]. However, the same cannot be applied to rigid structures like silica-based, polymeric, and metallic nanomedicines. If the required size based on the filter is not met, the process may cause loss of substantial active ingredient of the drug. Aseptic manufacturing is always used as a last resort owing to its multistep and complex nature to attain a sterile manufacturing environment.
Environmental Safety: Money at the Cost of Health
The most investigated issue for the manufacture of nanomedicines is environmental safety. The handling of drugs, their carriers or coatings in the dry form at the nanometre size scale may demand special caution due to the distribution of nanoparticles in the air as aerosols. Some nanomedicines during manufacturing stage are capable of penetrating the skin barrier, which provides potential risk to the engineers, workers and employees involved in the production, manufacturing and packaging of nanomedicines [72]. The deposition of such nanoparticles in the lungs can cause pulmonary toxicities [73]. The neurotoxicological analysis and various toxicity assessment assays performed on model organisms like zebrafish, Drosophila melanogaster and cell lines using different forms of nanoparticles are clear indicative of need of these tests before nanomedicines are released in the market [74,75,76]. The aerosolization of solutions should be avoided during solution preparation to prevent this undesired exposure. For this matter, the preparation of nanomedicines should be favourably done in liquid environment, similar to conventional pharmaceutical products, in order to reduce the burden of unintentionally released by nano-sized aerosols. The graveness of the issue is still vaguely understood to take firm authoritative actions.
Deranged Government Regulations
The FDA set the ball rolling regarding regulatory concerns of nanomaterials through a specific guidance document of August 2016. Though it addressed the general regulations for all nanomaterial products related to food ingredients, cosmetics, and animal feedstuff, but no special attention was paid towards nanomedicines. The draft guidance published by FDA in December 2017 recognized the high risk associated with nanopharmaceuticals which is likely to ‘exhibit clinically significant changes in exposure, safety, and/or effectiveness relative to the referenced products’ [15].
While the main regulatory bodies, US Food and Drug Administration (FDA), European Medicine Agency (EMA) and Therapeutic Goods and Administration, Australia (TGA) recognize the functional and compositional complexity of nanomedicines, the three do not follow the common tests and procedures while evaluating their adequate efficiency and safety. The compulsory parameters considered by the top regulatory authorities are listed in Table 2. The most important obstacle in regulation of nanomedicines is the lack of an agreed/consistent terminology and categorization of nanomedicines which complicates the communication process amongst the different regulatory bodies [77, 78]. The FDA categorizes its regulations into different groupings such as drugs, medical devices, biologic agents and blood products. The guideline for categorization of pharmaceutical agents relies on the ‘primary mode of action’. In cases of unclear or not entirely obvious primary mode of action, the discretion to decide the group of pharmaceutical product lies with the Office of Combination Products. For example, prosthetic bone cement equipped with nanotherapeutics or nanomaterials may fall into the categories of both devices and drugs.
Another FDA regulation that recently came under scrutiny is the premarket notification 510(k) which exempts certain medical devices intended for human use from Premarket Approval Application (PAA). It has paved a potential pathway for the release of unsatisfactory nanomedicines which need not go through arduous processes for approval. There is a long list of nanopharmaceutical products which were legitimately approved under 510(k) process [79, 80]. These products included dental composites, bone substitutes, medical dressings, tissue scaffolds, in vitro assays, device coatings and dialysis filters [81]. For instance, CarriGen, Ostim, NanOss approved through 510(k) are being used as bone substitutes for more than a decade now. The same applies to dental composites like Filtek, Nano-Bond; medical dressings like Acticoat and tissue scaffolds like TiMESH. The Dialysis Filter, Fresenius Polysulfone® and Helixone® have a market span of more than 20 years [81].
According to the recently issued first draft guidelines by The Ministry of Science and Technology, India [82], the evaluation of nanomedicine and nanopharmaceutical primarily concerns the engineered nanomaterials only. The document does not take into account the in vitro diagnostics, cell-based therapy products, tissue-engineered products and medical devices [65].
The absence of clearly defined tests and procedures that need to be cleared by novel nano-based drugs before applying for approval throws yet another challenge for nanopharmaceutical companies. Where each critical element should be checked for environmental risks and public hazards, it is not only the pristine form that needs assessment. The biological interaction of nanomaterials at developmental, manufacturing, therapeutic and disposal stage needs to be distinctly analysed for a thorough investigation.
The regulatory agencies need to be harmonized to urgently streamline approval processes and develop a comprehensive list of tests that covers the whole gamut of nanomedicine’s characterization, toxicological issues and pharmacology. The components, parameters and spatial composition interplays to decide the pharmokinetic response, safety profile and overall behaviour of nanoparticles [83].
Therefore, a fine balance needs to be achieved to ensure the quality and the safety of nanomedicines without the threat of over-regulation. This can hold back or progressively worsen the growth of innovative nanopharmaceutical products in the market by consuming a significant portion of the span of a patent and/or escalating the costs for achieving regulatory approvals.
Intellectual Property Rights: Prioritize Patients or Patents?
Though the academic research laboratories are constantly making extensive advances in nanomedicine, the question is to what extent these discoveries are being brought into the market through patents and licences issued to the pharmaceutical companies? There is not much data to precisely determine the share of nanodrugs in the total revenue of the medicinal companies. As per the classification of big pharma’s patent portfolio in 2015, the proportion of nanotech patents applied since 1994 accounted for only 0.3% in contrast to biotechnology patents ranging between 20 and 30% of the total share [84]. Now, in the face of being called the giant leap for technology, there is no place for nanotech amongst the top 40 most frequented IPC codes. One of the reasons for the small proportion of the patents may be the overlapping of nanotechnology with seven distinct applicative classes namely, agriculture and foodstuffs, medical sciences, physical or chemical processes or materials, organic chemistry, organic compounds, biochemistry and instruments (measuring, optical and computing) [85,86,87]. Being a young technology of a market career spanning 2–3 decades, nanotech cannot be termed as ‘stand-alone’ technology. The co-development of these domains also limits the number of patents being filed in the nanotech domain.
Nanomedicines acting as the bridge between medicine and medical applications has three broad classes for attaining Intellectual Property rights: for encapsulated nanodrug, for carrier technology and for the characteristics of drug and carrier together. Regardless of the straightforward categorization, the situation of patents in nanomedicines is very complex [88]. There are instances of incorporating existing drugs in a novel nanocarrier or existing drug with existing carrier technology used for a new biomedical use. The complicated drug delivery systems may use a nanocarrier, coatings or target ligands of one company whereas the therapeutic drug of another. It paves the way for the requirement of cross licensing, negotiations and arrangements, adding to the overall cost of development.
The share of nanobiotech-based products in the gross revenue earned by the pharmaceutical companies is not disclosed systematically. Based upon the annual reports of prominent medicinal giants, the share of individual nano-based drugs in the total earnings was analysed. For this matter, 65 pharma companies were considered in the initial data set but the unmethodical disclosure of revenue and sales from nanomedicines in quarterly and annual reports allowed only 15 products for complete analysis. The general trend represented in Table 3, and Fig. 2 suggests the hollowness of lucrative financial claims associated with nanomedicines.

Comparative analysis of annual sales of nanomedicines and gross sales of big pharmaceutical companies. Since data for the annual sales of 2019 for most pharmaceutical companies is yet to be published, the analysis considers the annual reports of the year 2018 to avoid any bias
Even the mega blockbuster biopharmaceutical drug HUMIRA, marketed by AbbVie, reported a decline of 5.6% in the revenue generated in the first quarter in 2018 [89]. HUMIRA is approved to treat various autoimmune diseases and its sale accounts for 61% of the total revenue generated by AbbVie. In 2018, the net revenues worldwide attained from HUMIRA accounted for USD 19.9 billion [90]. The coming of biosimilars in Europe and expiry of patent relating to the composition of matter of adalimumab (sold as HUMIRA) in the USA in December 2016 and most European countries in October 2018 may be the reason for this decline [90].
Ethical Challenges: Paving Way for Moral Blindness
The advancement of nanomedicines conjunctly arises the ethical questions associated with its use. The research stands on the building blocks of tension between the philosophical ethos of medical research concerning the development and production of effective diagnostic, therapeutic and preventive techniques versus the ethical imperative of the same enterprise that attempts to protect the well being and interests of the healthy individuals benefitting from breakthrough or innovations in research, patients, exposed workers and the public at large. The absence of firm ethical standards may lead to epistemic blindness and violation of human rights [91]. Since most of the human clinical trials of nanomedicines are carried out on patients at an advanced stage of disease with no other alternative therapy, the trials may offer no propitious effects or may even deteriorate the present state of the patient [92]. Therefore, it is always essential for the experts conducting the trials to attain an informed consent from the patients after explaining the procedures completely without giving false hopes or raising the hopes of the patients unjustifiable [93]. For all the ambivalence of stakeholders about disclosing the composition of medicine by mentioning the word ‘nano’, it is climacteric for the sake of patients’ confidence and moral righteousness to inform them about the use of nanoparticles in their treatment, even if it jeopardizes the likelihood of signing the consent forms [94]. Nevertheless, the overall ethical bindings for nanomedicinal trials remain the same as any other new therapy to be clinically evaluated. Therefore, where it should be obligatory to the industries and regulatory authorities to assess the benefit/risk ratio, there the discretion of participation and testing should lie with the patient. On that account, the stakeholders including scientists, industrialists, regulatory authorities and patients should strike a balance between being ignorant to the possible dangers of nanopharmaceuticals and being overcautious which may prohibit the development of these medicines altogether.
As for the socio-economic justice of nanomedicines, the accessibility and affordability of these products to all strata of the society should also be ensured. The average cost of conventional anticancer drug, doxorubicin costs around USD 62 to USD 162 per dose, whereas the nanoparticle coated doxorubicin called Doxil has an average cost of USD 5594 per dose [11]. On the same lines, the average cost of paclitaxel nanoformulation Abraxane is USD 5054 per dose, which is far beyond the capacity of patients of third world countries where health insurance is still a rarity. Another aspect of the problem is cost paid by insurance companies. According to a survey conducted in 2013, the health insurers of oncology patients pay USD 207,000 for an additional year of life in 2013 contrary to USD 54,100 paid in 1995 [95]. Anthem, which is the second largest health insurer in the United States, regards Abraxane as an overused product relative to its therapeutic value as paclitaxel, the parent drug of Abraxane has an average cost of USD 200 per dose [96]. Therefore, there is still a long way to go in the creation of an affordable and pharmacologically acclaimed market of nanomedicines for all sections of society. In addition to this, the industrialists should not attain excessive market control on the national or the international scale with unstringing IP laws and policies, but be subjugated to fair trade policies and stratified pricing programmes to promote economic justice. The international organizations should join hands to provide assistance and funding to developing countries to provide legit access to emerging products and better coverage of healthcare financing systems.
Nanomedicine Market: the Consolidation of Tumbling Stone
The present scenario is laden with worrying concern about the rising cost of the healthcare sector around the globe, inclusive of developed as well as developing countries. Moreover, the pathway for clinical translation is marred with stringent preclinical, clinical and market barriers as discussed above and represented in Fig. 3. Therefore, it is critical for governments and international regulatory agencies to acquire a deeper understanding of the efficiency of nanomedicines [97, 98]. The first step in the establishment of this market is experimental and analytical demonstration of over the edge benefits of nanopharmaceuticals compared with conventional formulations, which makes them worth their additional cost. By achieving this notion, the governments will acquire a free hand in issuing clear guidelines and projects to provide financial rewards for developing this market. Secondly, the field needs a comprehensive preclinical assessment of physicochemical characterization, efficacy, pharmacology and toxicological evaluations. The system also needs extensive in vitro and in vivo testing along with the implementation of alternative testing strategies and quality by design models. Moreover, there is a need for regulatory agencies to communicate in one language and make stringent guidelines for development, manufacture, testing and approval of all the new and innovative nanomedicines [99]. The manufacturing limitations and ethical challenges should also be addressed for long-term survival of these products. Finally, the quality of nanopharmaceutical products should be in line with defined critical quality attributes (CQAs) that not only satisfies the manufacturers and industrialists, but also patients and clinicians along with regulatory bodies for better clinical translation of nanomedicines. If we put simply, in order to persuade investors about the bombastic value of nanomedicines and create a wider horizon for its market, the intellectual property and regulatory authorities need to change their essence, formulate streamlined pathways and provide swift regulatory approvals with cautious and elaborate tests and analysis. In some cases, it is necessary to disburden the agencies, for now, take a step backward, forget the short-term benefits and reanalyse the situation, in order to formulate long-term plans with rationality.

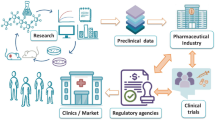
Stumbling blocks in the pathway for strategic clinical translation of nanomedicines, from discovery to delivery
Conclusion
The most prominent factor throughout the review is found to be the relative adolescence of the nanopharmaceutical sector in the market even with extensive research and preclinical studies being conducted. Although the regulatory authorities have given approval to a variety of nanomedicines, digital pills and nanodevices having significant technological advancements, they are only scratching the surface of the iceberg. The share of nanomedicines in healthcare sector occupies a limited part in pharmaceutical industries, both in terms of revenue and patents. Nanopharmaceutical research is predominantly a costly, lengthy and frightfully uncertain process, where it is the academic institutions that account for maximum innovations in this area. As a consequence, the expertise of the domain still lies with academia even after decades of hype created in nanomedicines. Faced with under skilled, inexperienced individuals and a looming threat of patent protection loss for many mega blockbuster drugs, the industries not only need to exploit and commercialize the academic research but also restock its pipeline with products and innovations promising potentially high returns. Moreover, the continued refinement and combination of the present technologies may lead to truly transformative capabilities envisioned for nanomedicine. But on the same side, the limitations in terms of design, manufacture, clinical, market and post market phase need to be carefully reflected upon to truly understand the efficacy of these drugs and their viability in the long run.
In the nutshell, the world is at a very pivotal juncture at this point in time. Based upon toxicological studies and long-term effects of these drugs, the decisions made today will have a long-lasting impact on our future generations. If the research analysts and scientific community miss the opportunity today to truly assess the balance between potential, utility, therapeutic efficacy and the underlying threats of using nanomedicines, we would have centuries to think and straighten out the chaos that we create now. The dream of healthy, peaceful and green earth should not be marred with counter drugs, water wars, pollution masks, inflating prices, downgraded economies and breakdown of social harmony.
Abbreviations
- FDA:
-
Food and Drug Administration
- SCID:
-
severe combined immunodeficiency
- CAGR:
-
compound annual growth rate
- PEG:
-
polyethylene glycol
- CDDS:
-
conventional drug delivery system
- NNI:
-
National Nanotechnology Initiative
- NIH:
-
National Institute of Health
- VC:
-
venture capital
- CMC:
-
critical micelle concentration
- PLGA:
-
poly(lactic-co-glycolic acid)
- MRI:
-
magnetic resonance imaging
- SSD:
-
species sensitivity distribution
- BGA:
-
band gap analysis
- QBD:
-
quality by design
- QSAR:
-
quantitative structure-activity relationship
- IND:
-
investigational new drug
- EMA:
-
European Medical Agency
- TGA:
-
Therapeutic Goods and Administration
- PAA:
-
premarket approval application
- IPC:
-
The International Patent Classification
- CQA:
-
critical quality attributes
References
Beltrán-Gracia E, López-Camacho A, Higuera-Ciapara I, Velázquez-Fernández JB, Vallejo-Cardona AA. Nanomedicine review: clinical developments in liposomal applications. Cancer Nanotechnol. Springer Vienna. 2019. https://doi.org/10.1186/s12645-019-0055-y.
Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016;33:2373–87. https://doi.org/10.1007/s11095-016-1958-5.
Lombardo D, Kiselev MA, Caccamo MT. Smart nanoparticles for drug delivery application: development of versatile nanocarrier platforms in biotechnology and nanomedicine. J Nanomater Hindawi Limited. 2019;2019.
Farjadian F, Ghasemi A, Gohari O, Roointan A, Karimi M, Hamblin MR. Nanopharmaceuticals and nanomedicines currently on the market: challenges and opportunities. Nanomedicine. 2019;14:93–126.
Iafisco M, Alogna A, Miragoli M, Catalucci D. Cardiovascular nanomedicine: the route ahead. Nanomedicine. Future Medicine Ltd. 2019:2391–4.
Formicola B, Cox A, Dal Magro R, Masserini M, Re F. Nanomedicine for the treatment of Alzheimer’s disease. J Biomed Nanotechnol. American Scientific Publishers. 2019:1997–2024.
Scherließ R. Future of nanomedicines for treating respiratory diseases. Expert Opin Drug Deliv, Taylor and Francis Ltd. 2019:59–68.
Ray L. Therapeutic Aspects of nanomedicines in stroke treatment. Adv Pathophysiol Cereb Stroke. Springer Singapore. 2019:139–53.
Meng T, Kulkarni V, Simmers R, Brar V, Xu Q. Therapeutic implications of nanomedicine for ocular drug delivery. Drug Discov Today. Elsevier Ltd. 2019:1524–38.
Agarwal V, Bajpai M, Sharma A. Patented and approval scenario of nanopharmaceuticals with relevancy to biomedical application, manufacturing procedure and safety aspects. Recent Pat Drug Deliv Formul. Bentham Science Publishers Ltd. 2018;12:40–52.
Global healthcare nanotechnology (nanomedicine) market—segmented by product type, application, marker type and geography—growth, trends and forecasts (2018–2023). 2018 [cited 2020 Feb 6]. Available from: https://www.marketresearch.com/Mordor-Intelligence-LLP-v4018/Global-Healthcare-Nanotechnology-Nanomedicine-Segmented-11528914/
Bregoli L, Movia D, Gavigan-Imedio JD, Lysaght J, Reynolds J, Prina-Mello A. Nanomedicine applied to translational oncology: a future perspective on cancer treatment. Nanomed Nanotechnol Biol Med. Elsevier Inc. 2016:81 103.
Halappanavar S, Vogel U, Wallin H, Yauk CL. Promise and peril in nanomedicine: the challenges and needs for integrated systems biology approaches to define health risk. Wiley Interdiscip Rev Nanomed Nanobiotechnol Wiley-Blackwell. 2018;10.
Bremer-Hoffmann S, Halamoda-Kenzaoui B, Borgos SE. Identification of regulatory needs for nanomedicines. J Interdiscip Nanomed. 2018;3:4–15 [cited 2020 Feb 6]; http://doi.wiley.com/10.1002/jin2.34.
U.S. Department of Health and Human Services Food and Drug Administration. Guidance for industry on drug products, including biological products, that contain nanomaterials. Fda. 2017; Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM588857.pdf
Emily M, Ioanna N, Scott B, Beat F. Reflections on FDA draft guidance for products containing nanomaterials: is the abbreviated new drug application (ANDA) a suitable pathway for nanomedicines? AAPS J. Springer New York LLC; 2018.
Degors IMS, Wang C, Rehman ZU, Zuhorn IS. Carriers break barriers in drug delivery: endocytosis and endosomal escape of gene delivery vectors. Acc Chem Res. 2019 [cited 2020 Feb 6];52:1750–60. Available from: https://pubs.acs.org/doi/10.1021/acs.accounts.9b00177
Karimi M, Karimi M. Future perspectives and the global drug delivery systems market. Smart Intern Stimulus-Responsive Nanocarriers Drug Gene Deliv. IOP Publishing; 2015.
Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update. Bioeng Transl Med Wiley. 2019;4.
Bansal M, Kumar A, Malinee M, Sharma TK. Nanomedicine: diagnosis, treatment, and potential prospects. 2020. p. 297–331.
Soares S, Sousa J, Pais A, Vitorino C. Nanomedicine: principles, properties, and regulatory issues. Front. Chem. Frontiers Media S.A.; 2018.
Qiao Y, Wan J, Zhou L, Ma W, Yang Y, Luo W, et al. Stimuli-responsive nanotherapeutics for precision drug delivery and cancer therapy. Wiley Interdiscip Rev Nanomedicine Nanobiotechnology. 2019 [cited 2020 Feb 6];11:e1527. Available from: http://doi.wiley.com/10.1002/wnan.1527
Nassiri Koopaei N, Abdollahi M. Opportunities and obstacles to the development of nanopharmaceuticals for human use. DARU, J. Pharm. Sci. BioMed Central Ltd.; 2016. p. 1–6.
Venkatraman S. Has nanomedicine lived up to its promise? Nanotechnology. Institute of Physics Publishing; 2014.
Caster JM, Patel AN, Zhang T, Wang A. Investigational nanomedicines in 2016: a review of nanotherapeutics currently undergoing clinical trials. Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology. Wiley-Blackwell; 2017.
Bhattacharya I, Heatherington A, Barton J. Applying the best of oncology drug development paradigms to the non-malignant space. Drug Discov. Today. Elsevier Ltd; 2016. p. 1869–72.
Di Masi JA, Grabowski HG, Hansen RW. The cost of drug development. N. Engl. J Med Massachussetts Medical Society; 2015. p. 1972.
Bosetti R, Jones SL. Cost-effectiveness of nanomedicine: estimating the real size of nano-costs. Nanomedicine. 2019;14:1367–70.
NNI Supplement to the President’s 2020 Budget | Nano. 2019 [cited 2020 Feb 6]. Available from: https://www.nano.gov/2020budgetsupplement
Strategic inputs of the ETPN | ETPN. 2019 [cited 2020 Feb 6]. Available from: https://etp-nanomedicine.eu/about-etpn/european-funding/
Mission on Nano Science and Technology (Nano Mission) | Department Of Science & Technology. 2019 [cited 2020 Feb 6]. Available from: https://dst.gov.in/scientific-programmes/mission-nano-science-and-technology-nano-mission
Northfelt DW, Dezube BJ, Thommes JA, Levine R, Von Roenn JH, Dosik GM, et al. Efficacy of pegylated-liposomal doxorubicin in the treatment of AIDS-related Kaposi’s sarcoma after failure of standard chemotherapy. J Clin Oncol. 1997;15:653–9.
Alza to buy Sequus $580 million deal to bolster development of new cancer drugs - SFGate. 1998 [cited 2020 Feb 6]. Available from: https://www.sfgate.com/business/article/Alza-to-buy-Sequus-580-million-deal-to-bolster-3066262.php.
Johnson & Johnson Announces Completion of Merger with ALZA Corporation | Johnson & Johnson. 2001 [cited 2020 Feb 6]. Available from: https://johnsonandjohnson.gcs-web.com/news-releases/news-release-details/johnson-johnson-announces-completion-merger-alza-corporation/
bind-10k_20151231.htm. 2016 [cited 2020 Feb 6]. Available from: https://www.sec.gov/Archives/edgar/data/1385228/000156459016014829/bind-10k_20151231.htm
Trouble brewing at Bind Therapeutics as major cuts announced | FierceBiotech. 2016 [cited 2020 Feb 6]. Available from: https://www.fiercebiotech.com/r-d/trouble-brewing-at-bind-therapeutics-as-major-cuts-announced
BIND Therapeutics Announces Completion of Collaboration Agreement with Amgen | FierceBiotech. 2014 [cited 2020 Feb 6]. Available from: https://www.fiercebiotech.com/biotech/bind-therapeutics-announces-completion-of-collaboration-agreement-amgen
Pfizer Wins Bid; Buys BIND Therapeutics Bankruptcy Assets for $40 Million | BioSpace. 2016 [cited 2020 Feb 6]. Available from: https://www.biospace.com/article/pfizer-wins-bid-buys-bind-therapeutics-bankruptcy-assets-for-40-million−/.
He H, Liu L, Morin EE, Liu M, Schwendeman A. Survey of clinical translation of cancer nanomedicines—lessons learned from successes and failures. Acc Chem Res. 2019 [cited 2020 Feb 6];52:2445–61. Available from: https://pubs.acs.org/doi/10.1021/acs.accounts.9b00228
Here’s Why Arrowhead Pharmaceuticals Lost as Much as 14.2% Today | The Motley Fool. 2020 [cited 2020 Feb 6]. Available from: https://www.fool.com/investing/2020/01/21/heres-why-arrowhead-pharmaceuticals-lost-as-much-a.aspx
MM-302 Falls Short in Phase II HER2+ Breast Cancer Trial. 2016 [cited 2020 Feb 6]. Available from: https://www.onclive.com/web-exclusives/mm302-falls-short-in-phase-ii-her2-breast-cancer-trial
Merrimack Discontinues Development of MM-310 | Merrimack Pharmaceuticals. 2019 [cited 2020 Feb 6]. Available from: http://investors.merrimack.com/news-releases/news-release-details/merrimack-discontinues-development-mm-310
Merrimack Stockholders Approve Sale of ONIVYDE® and Generic Version of DOXIL® to Ipsen for Up to $1.025 Billion | Merrimack Pharmaceuticals. 2017 [cited 2020 Feb 6]. Available from: http://investors.merrimack.com/news-releases/news-release-details/merrimack-stockholders-approve-sale-onivyder-and-generic-version
Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012;14:282–95.
Azhdarzadeh M, Saei AA, Sharifi S, Hajipour MJ, Alkilany AM, Sharifzadeh M, et al. Nanotoxicology: advances and pitfalls in research methodology. Nanomedicine. 2015;10:2931–52.
Moein Moghimi S, Simberg D, Skotland T, Yaghmur A, Christy Hunter A. The interplay between blood proteins, complement, and macrophages on nanomedicine performance and responses. J. Pharmacol Exp Ther. American Society for Pharmacology and Experimental Therapy; 2019. p. 581–92.
Faria M, Björnmalm M, Thurecht KJ, Kent SJ, Parton RG, Kavallaris M, et al. Minimum information reporting in bio–nano experimental literature. Nat Nanotechnol Nature Publishing Group. 2018;13:777–85.
Teleanu DM, Chircov C, Grumezescu AM, Teleanu RI. Neurotoxicity of nanomaterials: an up-to-date overview. Nanomaterials. 2019;9:1–14.
Gajewicz A, Puzyn T, Odziomek K, Urbaszek P, Haase A, Riebeling C, et al. Decision tree models to classify nanomaterials according to the DF4nanoGrouping scheme. Nanotoxicology. Taylor and Francis Ltd. 2018;12:1–17.
Ganguly P, Breen A, Pillai SC. Toxicity of nanomaterials: exposure, pathways, assessment, and recent advances. ACS Biomater Sci Eng American Chemical Society. 2018;4:2237–75.
Girelli D, Ugolini S, Busti F, Marchi G, Castagna A. Modern iron replacement therapy: clinical and pathophysiological insights. Int J Hematol Springer Tokyo. 2018;107:16–30.
Manoukian G, Hagemeister F. Denileukin diftitox: a novel immunotoxin. Expert Opin Biol Ther. 2009:1445–51.
Schwab CL, English DP, Roque DM, Pasternak M, Santin AD. Past, present and future targets for immunotherapy in ovarian cancer. Immunotherapy. 2014 [cited 2020 Feb 6];6:1279–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25524384.
Farjadian F, Moradi S, Hosseini M. Thin chitosan films containing super-paramagnetic nanoparticles with contrasting capability in magnetic resonance imaging. J Mater Sci Mater Med. Springer New York LLC; 2017;28.
Ventola CL. Progress in nanomedicine: approved and investigational nanodrugs. P T Medi Media USA Inc; 2017;42:742–55.
Accomasso L, Cristallini C, Giachino C. Risk assessment and risk minimization in nanomedicine: a need for predictive, alternative, and 3Rs strategies. Front Pharmacol. 2018;9:1–7.
Jang HL, Zhang YS, Khademhosseini A. Boosting clinical translation of nanomedicine. Nanomedicine. 2016;11:1495–7.
Li J, Qiao Y, Wu Z. Nanosystem trends in drug delivery using quality-by-design concept. J. Control. Release. Elsevier B.V.; 2017. p. 9–18.
Pallagi E, Ambrus R, Szabó-Révész P, Csóka I. Adaptation of the quality by design concept in early pharmaceutical development of an intranasal nanosized formulation. Int J Pharm Elsevier. 2015;491:384–92.
Agrahari V, Hiremath P. Challenges associated and approaches for successful translation of nanomedicines into commercial products. Nanomedicine. Future Medicine Ltd.; 2017. p. 819–23.
Barenholz Y. Doxil® - The first FDA-approved nano-drug: Lessons learned. J. Control. Release. 2012. p. 117–34.
Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release. Elsevier; 2015. p. 138–57.
Leong HS, Butler KS, Brinker CJ, Azzawi M, Conlan S, Dufés C, et al. On the issue of transparency and reproducibility in nanomedicine. Nat. Nanotechnol. Nat Publ Group; 2019. p. 629–635.
Tinkle S, McNeil SE, Mühlebach S, Bawa R, Borchard G, Barenholz YC, et al. Nanomedicines: addressing the scientific and regulatory gap. Ann N Y Acad Sci. 2014 [cited 2020 Feb 6];1313:35–56. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24673240.
Marques MRC, Choo Q, Ashtikar M, Rocha TC, Bremer-Hoffmann S, Wacker MG. Nanomedicines - Tiny particles and big challenges. Adv. Drug Deliv. Rev. Elsevier B.V.; 2019. p. 23–43.
Singh MK, Kuncha M, Nayak VL, Sarma AVS, Kumar MJM, Chauhan AS, et al. An innovative in situ method of creating hybrid dendrimer nano-assembly: an efficient next generation dendritic platform for drug delivery. Nanomedicine Nanotechnology, Biol Med. Elsevier Inc.; 2019;21.
Cong Y, Ji L, Gao Y-J, Liu F-H, Cheng D-B, Hu Z, et al. Microenvironment-induced in situ self-assembly of polymer-peptide conjugates that attack solid tumors deeply. Angew Chemie Int Ed. 2019 [cited 2020 Feb 6];58:4632–7. Available from: http://doi.wiley.com/10.1002/anie.201900135
Liang C, Yan X, Zhang R, Xu T, Zheng D, Tan Z, et al. Enhanced cellular uptake and nuclear accumulation of drug-peptide nanomedicines prepared by enzyme-instructed self-assembly. J Control Release. Elsevier B.V.; 2020;317:109–17.
Wu LP, Wang D, Li Z. Grand challenges in nanomedicine. Mater Sci Eng C. Elsevier; 2020;106:110302. Available from: https://doi.org/10.1016/j.msec.2019.110302.
Rafael D, Andrade F, Martinez-Trucharte F, Basas J, Seras-Franzoso J, Palau M, et al. Sterilization procedure for temperature-sensitive hydrogels loaded with silver nanoparticles for clinical applications. Nanomater (Basel, Switzerland). 2019 [cited 2020 Feb 6];9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30845683.
Crommelin DJA, van Hoogevest P, Storm G. The role of liposomes in clinical nanomedicine development. What now? Now what? J Control Release. Elsevier B.V.; 2020;318:256–63.
Erkekoglu P, Kocer-Gumusel B. Toxicity assessment of nanopharmaceuticals. Inorg Fram as Smart Nanomedicines. William Andrew; 2018. p. 565–603.
Li G, Liang L, Yang J, Zeng L, Xie Z, Zhong Y, et al. Pulmonary hypofunction due to calcium carbonate nanomaterial exposure in occupational workers: a cross-sectional study. Nanotoxicology Taylor and Francis Ltd. 2018;12:571–85.
Khatri M, Bello D, Martin J, Bello A, Gore R, Demokritou P, et al. Chronic upper airway inflammation and systemic oxidative stress from nanoparticles in photocopier operators: mechanistic insights. NanoImpact. Elsevier B.V.; 2017;5:133–45.
Kaur J, Khatri M, Puri S. Toxicological evaluation of metal oxide nanoparticles and mixed exposures at low doses using zebra fish and THP1 cell line. Environ Toxicol. 2019 [cited 2020 Feb 6];34:375–87. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30548797.
Sood K, Kaur J, Singh H, Kumar Arya S, Khatri M. Comparative toxicity evaluation of graphene oxide (GO) and zinc oxide (ZnO) nanoparticles on Drosophila melanogaster. Toxicol Reports. Elsevier Inc.; 2019;6:768–81.
Paradise J. Regulating nanomedicine at the food and drug administration. AMA J Ethics. American Medical Association; 2019;21:347–55.
Tambe V, Maheshwari R, Chourasiya Y, Choudhury H, Gorain B, Tekade RK. Clinical aspects and regulatory requirements for nanomedicines. Basic Fundam Drug Deliv. Elsevier Inc.; 2019. Available from: https://doi.org/10.1016/B978-0-12-817909-3.00018-2
Van Norman GA. Drugs, devices, and the FDA: part 2: an overview of approval processes: FDA approval of medical devices. JACC Basic to Transl Sci Elsevier Inc. 2016;1:277–87.
Jones AAD, Mi G, Webster TJ. A status report on FDA approval of medical devices containing nanostructured materials. Trends Biotechnol. Elsevier Ltd; 2019. p. 117–20.
Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine Nanotechnology, Biol Med. Elsevier Inc.; 2013;9:1–14. Available from: https://doi.org/10.1016/j.nano.2012.05.013.
Welfare F. Guidelines for evaluation of nanopharmaceuticals in India guidelines for evaluation of Nanopharmaceuticals in India. 2019;
Choi YH, Han HK. Nanomedicines: current status and future perspectives in aspect of drug delivery and pharmacokinetics. J. Pharm. Investig. Springer Netherlands; 2018. p. 43–60.
Würmseher M, Firmin L. Nanobiotech in big pharma: a business perspective. Nanomedicine. 2017;12:535–43.
Patenting nanotechnology: exploring the challenges. 2011 [cited 2020 Feb 6]. Available from: https://www.wipo.int/wipo_magazine/en/2011/02/article_0009.html
Windheim J von, Myers B. A lab-to-market roadmap for early-stage entrepreneurship. Transl Mater Res. IOP Publishing; 2014;1:016001.
Bosetti R, Vereeck L. The impact of effective patents on future innovations in nanomedicine. Pharm Pat Anal. 2012 [cited 2020 Feb 6];1:37–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24236712.
Brougher J, Linnik KM. Patents or patients: who loses? Nat Biotechnol. Nature publishing group; 2014;32:877–80. Available from: https://doi.org/10.1038/nbt.3005.
Say goodbye to AbbVie’s ever-growing Humira sales: the biosim decline has officially begun | FiercePharma. 2019 [cited 2020 Feb 6]. Available from: https://www.fiercepharma.com/pharma/abbvie-looks-to-new-launches-as-humira-starts-to-crack
AbbVie. AbbVie Annual Report 2018. 2018;
Solbakk JH, Vidal SM. Clinical research in resource-poor settings. Handb Glob Bioeth. Springer Netherlands; 2014. p. 527–50.
Satalkar P, Elger BS, Hunziker P, Shaw D. Challenges of clinical translation in nanomedicine: a qualitative study. Nanomedicine Nanotechnology, Biol Med. Elsevier Inc.; 2016;12:893–900.
Allon I, Ben-Yehudah A, Dekel R, Solbakk JH, Weltring KM, Siegal G. Ethical issues in nanomedicine: tempest in a teapot? Med Heal Care Philos Springer Netherlands; 2017;20:3–11.
Satalkar P, Elger BS, Shaw D. Naming it “nano”: expert views on “nano” terminology in informed consent forms of first-in-human nanomedicine trials. Nanomedicine (Lond). 2016 [cited 2020 Feb 6];11:933–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26979933.
U.S. cancer doctors drop pricey drugs with little or no effect - Reuters. 2015 [cited 2020 Feb 6]. Available from: https://www.reuters.com/article/us-usa-healthcare-cancer-insight/u-s-cancer-doctors-drop-pricey-drugs-with-little-or-no-effect-idUSKCN0S20DG20151008
Weissig V, Guzman-Villanueva D. Nanopharmaceuticals (part 2): products in the pipeline. Int J Nanomedicine. 2015 [cited 2020 Feb 6];10:1245–57. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25709446.
Zingg R, Fischer M. The consolidation of nanomedicine. Wiley Interdiscip Rev Nanomedicine Nanobiotechnology. 2019 [cited 2020 Feb 6];11. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wnan.1569
Agrahari V, Agrahari V. Facilitating the translation of nanomedicines to a clinical product: challenges and opportunities. Drug Discov. Today. Elsevier Ltd; 2018. p. 974–991.
Zamboni WC, Torchilin V, Patri AK, Hrkach J, Stern S, Lee R, et al. Best practices in cancer nanotechnology: perspective from NCI nanotechnology alliance. Clin Cancer Res. 2012 [cited 2020 Feb 6];18:3229–41. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22669131.
Acknowledgements
Dr. Madhu Khatri acknowledges Wellcome Trust/DBT Alliance for Early Career fellowship. Dr. Neha Bhardwaj acknowledges Department of Science & Technology (DST) for the INSPIRE FACULTY grant (Reg. no. IFA18-LSPA 127).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Nanomedicines are extensively researched for their highly claimed efficacy, biocompatibility and unexplored potential.
• The clinical translation of nanopharmaceuticals from laboratory to market undergoes many challenges on their pathway.
• Major hurdles restricting the market dominance of nanomedicines includes limited investments by pharmaceutical, toxicological assays, biocompatibility risks, scale-up issues, regulatory limitations, etc.
• Comparative analysis of annual reports of big pharmaceutical companies for gross sales and revenue contributed by nanomedicines.
Recommendations to improve the research, development, manufacture and sales of nanopharmaceuticals.
Rights and permissions
About this article

Cite this article
Kad, A., Pundir, A., Arya, S.K. et al. An Elucidative Review to Analytically Sieve the Viability of Nanomedicine Market. J Pharm Innov 17, 249–265 (2022). https://doi.org/10.1007/s12247-020-09495-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-020-09495-5