
Abstract
Acute promyelocytic leukemia (APL) is an uncommon subtype of acute myelogenous leukemia characterized by the proliferation of blasts with distinct morphology, a specific balanced reciprocal translocation t(15;17), and life-threatening hemorrhage caused mainly by enhanced fibrinolytic-type disseminated intravascular coagulation (DIC). The introduction of all-trans retinoic acid (ATRA) into anthracycline-based induction chemotherapy regimens has dramatically improved overall survival of individuals with APL, although hemorrhage-related death during the early phase of therapy remains a serious problem. Moreover, population-based studies have shown that the incidence of early death during induction chemotherapy is nearly 30 %, and the most common cause of death is associated with hemorrhage. Thus, development of a novel treatment strategy to alleviate abnormal coagulation in APL patients is urgently required. Recombinant human soluble thrombomodulin (rTM) comprises the active extracellular domain of TM, and has been used for treatment of DIC since 2008 in Japan. Use of rTM in combination with remission induction chemotherapy, including ATRA, produces potent resolution of DIC without exacerbation of bleeding tendency in individuals with APL. This review article discusses the pathogenesis and features of DIC caused by APL, as well as the possible anticoagulant and anti-leukemic action of rTM in APL patients.
Similar content being viewed by others

Introduction
Acute promyelocytic leukemia (APL) constitutes approximately 10 % of all cases of adult acute myelogenous leukemia (AML) and is characterized by a specific balanced reciprocal translocation t(15;17), generating promyelocytic leukemia–retinoic acid receptor α (PML–RARα) fusion transcripts that impair signaling mediated by RARα [1, 2]. The most important clinical feature of APL is life-threatening hemorrhage, which is caused mainly by enhanced fibrinolytic-type disseminated intravascular coagulation (DIC). Although the true incidence of DIC in APL patients is unknown, a recent epidemiological survey conducted in Taiwan found that 90 (77.6 %) of 116 APL patients developed overt DIC [3].
Prior to the ATRA era, early mortality related to hemorrhagic complications was extremely high [4–8] (Table 1). For example, early death during the first course of chemotherapy was observed in 47 % of APL patients (n = 57) treated with combination chemotherapy with daunorubicin (DNR) and cytarabine (AraC). Approximately, 41 % of early deaths were related to hemorrhagic events, with intracranial hemorrhage being the most common cause of death (73 %) [6]. Incorporation of all-trans retinoic acid (ATRA) and, more recently, arsenic trioxide (ATO), into induction chemotherapy has revolutionized the treatment of individuals with APL, with 90–95 % of newly diagnosed APL patients achieving complete remission and over 85 % of patients surviving for longer than 5 years [9–23]. Despite the incorporation of ATRA, hemorrhage remains the major cause of early death (4.5 %); other causes of early death include infection (1.9 %) and differentiation syndrome (1.2 %) [20] (Table 1). Moreover, population-based studies have shown that the early death rate during induction chemotherapy remains extremely high (30 %), and the most common cause of death is associated with hemorrhage [24–26] (Table 1). Thus, development of a novel treatment strategy to alleviate coagulopathy in APL patients is urgently required.
Recombinant human soluble thrombomodulin (rTM) comprises the active, extracellular domain of thrombomodulin (TM) and inactivates coagulation by binding to thrombin [27, 28]. In addition, the thrombin–rTM complex activates protein C to produce activated protein C (APC), which inactivates factors VIIIa and Va in the presence of protein S, further inhibiting thrombin formation [29]. The use of rTM for the treatment of DIC was approved in Japan in 2008, and is effective for the management of DIC complicated by a variety of underlying diseases [30–36]. rTM possesses anti-inflammatory and cytoprotective effects [37–39], and the use of rTM significantly improved survival of mechanically ventilated patients with severe sepsis when compared with control patients who did not receive rTM [30]. Multifunctional rTM has recently come into the global spotlight as a novel agent for the management of DIC [40, 41].
Clinical manifestation of DIC in APL patients
DIC complicated by APL is characterized by exaggerated fibrinolysis and life-threatening hemorrhage. Hemorrhagic death frequently occurred within the first week after initiation of induction chemotherapy, and was nearly exclusively caused by intracranial and pulmonary hemorrhages (incidence of 65 and 32 %, respectively). These data come from the Programa de Estudio y Tratamiento de las Hemopatias Malignas (PETHEMA) group that carefully evaluated the cause of induction failure in 732 newly diagnosed APL patients who were treated with a combination of ATRA and idarubicin [16]. High blast count (>30 × 109/L) was identified as a predictive factor for hemorrhagic death by PETHEMA as well as by the Gruppo Italiano per le Malattie Ematologiche dell’Adulto (GIMEMA) group that evaluated early hemorrhagic death in 622 consecutive APL patients treated with ATRA either alone (n = 499) or in combination with idarubicin (n = 123) [13].
Notably, approximately 5 % of APL patients developed thrombotic events, including deep venous thromboses, cerebral strokes, pulmonary emboli, and myocardial infarction, during induction chemotherapy with ATRA and idarubicin [42, 43]. Initiation of therapy with ATRA rapidly corrects hyperfibrinolysis; however, normalization of thrombotic markers, such as prothrombin fragment 1 + 2 (F1 + 2) and thrombin antithrombin complex (TAT), does not occur until 2 weeks after treatment with ATRA [44]. It may be that ATRA impairs the balance between coagulation and fibrinolysis and thereby causes a hypercoagulable state. It should also be noted that severe thrombotic complications occurred in six of 26 patients who died before initiation of chemotherapy [43]. This PETHEMA study identified a low level of fibrinogen and microgranular morphology (M3 variant) as an independent prognostic factor for thrombosis [43]. Importantly, prophylactic use of tranexamic acid did not impact hemorrhagic mortality, but it did tend to increase the incidence of thrombotic complications [43].
Pathogenesis of DIC caused by APL
Hypercoagulability
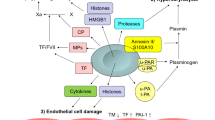
Plasma markers of clotting activation, such as TAT, F1 + 2, and fibrinopeptide A (FPA), are markedly elevated in APL patients [44–46], indicating the presence of hypercoagulability. There are three different types of procoagulants aberrantly produced by APL cells: tissue factor (TF), cancer procoagulant (CP), and microparticles (MPs) (Fig. 1). TF, a transmembrane glycoprotein, forms a complex with factor VII (FVII) to activate coagulation factor X (FX), which generates thrombin. As APL cell line NB4 underwent apoptosis after exposure to cytotoxic agents, thrombin generation was augmented in association with enhanced TF activity. The exteriorization of phosphatidylserine on the surface of APL cells during apoptosis allows interaction with the extracellular domain of TF, leading to its activation [47]. These observations are clinically relevant; DIC is exacerbated in APL patients when tumor lysis occurs after initiation of induction chemotherapy with cytotoxic agents, such as anthracycline and AraC. CP, a cysteine proteinase procoagulant produced from fetal and malignant cells, directly activates FX and generates thrombin in the absence of FVII. Among the different cytological subtypes of AML, the activity of CP was greatest in APL cells [48, 49]. MPs might also be involved in hypercoagulability in APL patients. MPs are released from APL cells, as evidenced by their expression of CD33 cell surface antigen and TF. The number of CD33+ MPs strongly correlated with leukocyte counts and plasma levels of D-dimer in APL patients [50]. APL cell-derived MPs decreased coagulation time and increased thrombin generation in a TF-dependent manner [50]. Exposure of leukemia cells to anthracycline increased the release of TF+ MPs in vitro [51]. This may account for the exacerbation of DIC during remission induction chemotherapy.

1 Hypercoagulability. APL cells express TF, CP and MPs, which activate the coagulation cascade. 2 Hyperfibrinolysis. APL cells produce t-PA, u-PA and u-PAR, which activate plasminogen. APL cells aberrantly express annexin II on their cell surface, which mediates conversion of plasminogen to plasmin. Elastases produced by APL cells may cleave fibrinogen and degrade fibrinolytic inhibitors, resulting in hyperfibrinolysis. 3 Endothelial cell damage. APL cells produce various types of inflammatory cytokine, including IL-1β, IL-6 and TNF-α, all of which cause endothelial cell damage and downregulate expression of TM in parallel with upregulation of TF and PAI-1 on the cell surface of endothelial cells, resulting in hypercoagulability. TF, tissue factor; CP, cancer procoagulant; MPs, microparticles; t-PA, tissue-type plasminogen activator; u-PA, urokinase-type plasminogen activator; u-PAR, u-PA receptor; IL-1β, interleukin-1β; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; TM, thrombomodulin; PAI-1, plasminogen activator inhibitor-1
APL cells produce a variety of inflammatory cytokines, including interleukin (IL)-1β, IL-6 and tumor necrosis factor-α (TNF-α) [52]. These cytokines cause endothelial cell damage and downregulate the expression of TM in parallel with upregulation of TF and plasminogen activator inhibitor-1 (PAI-1) on the cell surface of endothelial cells, resulting in hypercoagulability [53, 54] (Fig. 1).
Hyperfibrinolysis
Laboratory findings of DIC in patients with APL are characterized by marked hypofibrinogenemia and an increase in ratio of fibrin/fibrinogen degradation products (FDP)/D-dimer. Moreover, a decrease in the levels of plasminogen, α-2-antiplasmin and plasminogen activator inhibitor 1 (PAI-1) and an increase in the levels of urokinase-type plasminogen activator (u-PA) and plasmin/α-2-antiplasmin complex were noted in APL patients [55–58]. These observations may explain the exaggerated fibrinolysis in APL patients. Further studies have demonstrated that APL cells expressed several key mediators of plasmin generation, such as tissue-type plasminogen activator (t-PA), u-PA and its receptor, u-PAR. The u-PA/u-PAR complex enhances activation of cell-bound plasminogen [59, 60] (Fig. 1).
One of the most important factors responsible for exaggerated fibrinolysis in APL is the cell surface phospholipid-binding protein, annexin II (Fig. 1). Annexin II is expressed on endothelial cells and macrophages and acts as a cell surface receptor for plasmin and t-PA [61]. t-PA-mediated conversion of plasminogen to plasmin on annexin II results in a 60-fold increase in plasmin generation when compared with that in the fluid phase [62]. Intriguingly, APL cells express higher levels of annexin II and more efficiently generate plasmin than t(15;17) fusion gene-negative AML cells [63]. Importantly, exposure of APL cells to ATRA downregulates the expression of annexin II and inhibits plasmin activity in vitro [63].
Another molecule that may be related to hemorrhagic diatheses in APL is thrombin-activatable fibrinolysis inhibitor (TAFI). Thrombin protects fibrin clots from plasmin-mediated fibrinolysis via activation of this carboxypeptidase; thrombin bound to TM efficiently activates TAFI, which hampers fibrinolysis by removing C-terminal lysine residues on fibrin that are otherwise important for binding of plasminogen and t-PA, thereby efficiently generating plasmin [64, 65]. It has been suggested that TAFI may block exaggerated fibrinolysis in this manner. In fact, inhibition of TAFI activity by a carboxypeptidase inhibitor stimulated fibrinolysis in an animal model [66]. Notably, activity of TAFI was inhibited by 60 % in APL patients [67].
Granulocytic proteases, including elastases, are abundantly produced by APL cells. These proteases are thought to cleave fibrinogen and degrade fibrinolytic inhibitors, leading to augmented fibrinolysis [68, 69] (Fig. 1). However, the contribution of granulocytic proteases released by APL cells to hemorrhagic events is not clear; even after improvement of coagulopathy in APL patients in response to ATRA, plasma levels of elastases remained high [69]. In addition, recent studies comparing elastase-mediated fibrinolytic activity between sepsis-induced DIC and APL-related coagulopathy showed that levels of elastase-degraded fibrin (ogen) were significantly higher in sepsis patients [70].
Management of DIC caused by APL
Intensive supportive care consisting of fresh frozen plasma, fibrinogen, and/or cryoprecipitate and platelet transfusions to maintain the levels of fibrinogen and platelets above 100–150 mg/dL and 30–50 × 109/L, respectively, is strongly encouraged by European LeukemiaNet [71]. Of note, 18 (6.5 %) of 279 APL patients treated with an ATRA-containing regimen developed severe hemorrhage, and these patients received frequent transfusions to maintain the levels of fibrinogen and platelet count above 150 mg/dL and 30 × 109/L, respectively [18]. Only 40 and 71 % of patients achieved target levels of fibrinogen and platelet, respectively, at the onset of bleeding [18]. This observation suggests that intensive transfusion during remission induction chemotherapy is not sufficient to overcome coagulopathy. Therefore, novel treatments are clearly required to promptly correct abnormal coagulation in APL patients.
European LeukemiaNet does not recommend the use of heparin, tranexamic acid, or other anticoagulant or antifibrinolytic agents for the management of coagulopathy in APL patients, as the ability of these agents to reduce hemorrhagic risk was found to be questionable [71]. Only one retrospective study conducted in the UK found that heparin was clinically beneficial (Table 1); however, the dose of heparin and the treatment period varied between each patient, according to their physicians’ decisions. Also, the induction therapy regimen was not uniform in this retrospective study [7].
rTM was approved for the treatment of DIC in Japan in 2008. A phase III trial comparing the efficacy and safety of rTM and low-dose heparin showed that rTM significantly improved DIC that was associated with hematological malignancies or infections [72]. This clinical trial excluded almost all patients with APL, because concomitant use of ATRA was prohibited. Since 2008, the use of rTM has proved effective and safe in individuals with DIC and APL [73–75]. Use of rTM in combination with chemotherapy including ATRA rescued APL patients (n = 9) from DIC earlier than control patients (n = 8) who did not receive rTM [73]. No bleeding-related mortality was noted during induction chemotherapy in APL patients who received rTM [73]. Notably, severe hemorrhage requiring red blood cell transfusion at the time of diagnosis of APL was reduced after initiation of rTM [74]. Use of rTM in combination with ATO for relapse of APL promptly improved DIC without any adverse effect in one case report [75]. These observations suggest that rTM is a promising agent for the management of coagulopathy in APL patients, although the sample size in these studies is extremely small.
Structure and function of TM
TM is a glycosylated type I transmembrane molecule of 557 amino acids with multiple domains. Each domain possesses distinct properties. The molecule consists of an NH2-terminal lectin-like region followed by six tandem epidermal growth factor (EGF)-like structures, an o-glycosylation site-rich domain, a transmembrane domain, and a cytoplasmic tail domain [76] (Fig. 2). rTM comprises the extracellular domains of TM. TM is ubiquitously expressed on endothelial cells and binds to thrombin, forming a 1:1 complex via E45 repeats in an EGF-like domain and acting as an anticoagulant [28]. In addition, the thrombin–TM complex activates protein C to produce APC, which inactivates factors VIIIa and Va in the presence of protein S, thereby inhibiting further thrombin formation [29]. The minimum structure essential to generate APC is localized in E456 repeats of the EGF-like domain [77]. On the other hand, E3456 repeats are responsible for activation of TAFI, which acts as an anti-fibrinolytic [64, 65].

The structure and function of TM. The EGF-like domain of TM exerts anticoagulant effects by inactivating thrombin and indirectly inhibiting coagulation factor Va and VIIIa via APC. On the other hand, the E3456 repeats of the EGF-like domain are responsible for activation of TAFI, leading to anti-fibrinolysis. The lectin-like domain of TM possesses anti-inflammatory function. The E45 repeats of the EGF-like domain exert cytoprotective effects in an APC-independent manner. APC also exerts anti-inflammatory and cytoprotective effects. rTM comprises the functionary active extracellular domains of TM. TM, thrombomodulin; EGF, epidermal growth factor; TAFI, thrombin-activatable fibrinolysis inhibitor; APC, activated protein C; FVa, activated factor V; FVIIIa, activated factor VIII; rTM, recombinant human soluble thrombomodulin
TM possesses anti-inflammatory properties; the lectin-like domain of TM binds to and inactivates high-mobility group box 1 protein (HMGB1), a proinflammatory cytokine that stimulates production of inflammatory cytokines, such as IL-6 and TNF-α, via toll-like receptor 4 and that serves as a receptor for advanced glycation end products [37]. In addition, the lectin-like domain of TM inhibits lipopolysaccharide-induced cytokine production and adhesion of neutrophils to endothelial cells in association with suppression of extracellular signal-regulated kinase (ERK) and nuclear factor κ B (NF-κB) [38].
Intriguingly, TM exerts endothelial cytoprotective effects against inflammatory cytokines or calcineurin inhibitors via upregulation of myeloid cell leukemia sequence 1 (Mcl-1) proteins in a process that is mediated by the ERK signal transduction pathway [39]. This cytoprotective effect is also mediated by E45 repeats of the EGF-like domain, and is independent of APC [39]. Of note, TM also exerts anti-inflammatory and cytoprotective effects via APC-dependent mechanisms [78–81].
Possible action of rTM in APL cells
An in vitro study demonstrated that exposure of APL cells to rTM significantly downregulates levels of annexin II, resulting in a decrease in plasmin production [82]. rTM-induced downregulation of annexin II in APL cells was dependent on APC, but the precise mechanisms by which this occurs remain unknown. ATRA also inhibited plasmin production in association with downregulation of annexin II in APL cells [63]. Interestingly, when rTM was combined with ATRA, inhibition of plasmin production in APL cells was synergistically enhanced [82]. Remarkably, rTM alone was able to induce myeloid differentiation and growth arrest of APL cells in association with induction of CCAAT/enhanced binding protein ε, an essential nuclear transcription factor for myeloid differentiation in an APC-dependent mechanism [82]. Again, rTM synergized with ATRA to produce anti-APL effects [81]. ATRA increased TM levels in acute myeloid leukemia HL60 cells as they differentiated toward neutrophils [83]. ATRA can regulate the expression of TM via the RA response element located on the 5′-flanking region of this gene [84]. Thus, TM may, at least in part, mediate ATRA-induced myeloid differentiation of APL cells. ATRA also induced the expression of TM on the cell surface of endothelial cells [85]. ATRA may normalize abnormal coagulation via induction of TM on the endothelium and APL cells in individuals with APL.
A major clinical problem in addition to coagulopathy in APL patients after initiation of ATRA and/or ATO is differentiation syndrome (DS), formerly known as retinoic acid syndrome. DS is characterized by unexplained fever and acute respiratory distress with vascular capillary leakage [86–88]. The patho-etiology of DS remains to be fully elucidated, but the insults to the respiratory and vascular endothelium caused by cytokines released by differentiated myeloid cells are considered to be involved in the development of this potentially lethal syndrome [89]. rTM may counteract DS, as rTM successfully alleviated capillary leakage in individuals with engraftment syndrome and sinusoidal obstruction syndrome that developed after hematopoietic stem cell transplantation [35, 36].
Conclusions
Introduction of ATRA and ATO into remission induction chemotherapy for individuals with APL has dramatically improved clinical outcome of this fatal subtype of leukemia; however, the incidence of life-threatening hemorrhagic and thrombotic events in APL patients at the time of diagnosis and/or during induction chemotherapy is still higher than expected. Use of the anticoagulant, rTM, which has additional activity against exaggerated fibrinolysis, inflammation, and endothelial cell damage, is a promising treatment strategy to safely rescue APL patients from life-threatening coagulopathy. Further large cohort studies are clearly required to establish the safety and efficacy of rTM for management of coagulopathy in patients with APL.
References
Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92:2322–33.
Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391:815–8.
Chang H, Kuo MC, Shih LY, Dunn P, Wang PN, Wu JH, et al. Clinical bleeding events and laboratory coagulation profiles in acute promyelocytic leukemia. Eur J Haematol. 2012;88:321–8.
Bernard J, Weil M, Boiron M, Jacquillat C, Flandrin G, Gemon MF. Acute promyelocytic leukemia: results of treatment by daunorubicin. Blood. 1973;41(4):489–96.
Cunningham I, Gee TS, Reich LM, Kempin SJ, Naval AN, Clarkson BD. Acute promyelocytic leukemia: treatment results during a decade at memorial hospital. Blood. 1989;73:1116–22.
Cordonnier C, Vernant JP, Brun B, Heilmann MG, Kuentz M, Bierling P, Farcet JP. Acute promyelocytic leukemia in 57 previously untreated patients. Cancer. 1985;55:18–25.
Hoyle CF, Swirsky DM, Freedman L, Hayhoe FG. Beneficial effect of heparin in the management of patients with APL. Br J Haematol. 1988;68:283–9.
Rodeghiero F, Avvisati G, Castaman G, Barbui T, Mandelli F. Early deaths and anti-hemorrhagic treatments in acute promyelocytic leukemia. A GIMEMA retrospective study in 268 consecutive patients. Blood. 1990;75:2112–7.
Fenaux P, Castaigne S, Dombret H, Archimbaud E, Duarte M, Morel P, et al. All-transretinoic acid followed by intensive chemotherapy gives a high complete remission rate and may prolong remissions in newly diagnosed acute promyelocytic leukemia: a pilot study on 26 cases. Blood. 1992;80:2176–81.
Kanamaru A, Takemoto Y, Tanimoto M, Murakami H, Asou N, Kobayashi T, et al. All-trans retinoic acid for the treatment of newly diagnosed acute promyelocytic leukemia. Japan Adult Leukemia Study Group. Blood. 1995;85:1202–6.
Fenaux P, Le Deley MC, Castaigne S, Archimbaud E, Chomienne C, Link H, et al. Effect of all transretinoic acid in newly diagnosed acute promyelocytic leukemia. Results of a multicenter randomized trial. European APL 91 Group. Blood. 1993;82:3241–9.
Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337:1021–8.
Di Bona E, Avvisati G, Castaman G, Luce Vegna M, De Sanctis V, Rodeghiero F, et al. Early haemorrhagic morbidity and mortality during remission induction with or without all-trans retinoic acid in acute promyelocytic leukaemia. Br J Haematol. 2000;108:689–95.
Estey E, Thall PF, Pierce S, Kantarjian H, Keating M. Treatment of newly diagnosed acute promyelocytic leukemia without cytarabine. J Clin Oncol. 1997;15:483–90.
Fenaux P, Chastang C, Chevret S, Sanz M, Dombret H, Archimbaud E, et al. A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1999;94:1192–200.
de la Serna J, Montesinos P, Vellenga E, Rayón C, Parody R, León A. Causes and prognostic factors of remission induction failure in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and idarubicin. Blood. 2008;111:3395–402.
Asou N, Adachi K, Tamura J, Kanamaru A, Kageyama S, Hiraoka A, et al. All-trans retinoic acid therapy for newly diagnosed acute promyelocytic leukemia: comparison with intensive chemotherapy. The Japan Adult Leukemia Study Group (JALSG). Cancer Chemother Pharmacol. 1997;40(Suppl):S30–5.
Yanada M, Matsushita T, Asou N, Kishimoto Y, Tsuzuki M, Maeda Y, et al. Severe hemorrhagic complications during remission induction therapy for acute promyelocytic leukemia: incidence, risk factors, and influence on outcome. Eur J Haematol. 2007;78:213–9.
Lo-Coco F, Avvisati G, Vignetti M, Breccia M, Gallo E, Rambaldi A, Paoloni F. Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood. 2010;116:3171–9.
Sanz MA, Montesinos P, Rayón C, Holowiecka A, de la Serna J, Milone G. Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome. Blood. 2010;115:5137–46.
Mathews V, George B, Lakshmi KM, Viswabandya A, Bajel A, Balasubramanian P, et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: durable remissions with minimal toxicity. Blood. 2006;107:2627–32.
Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM, et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA. 2004;101:5328–35.
Hu J, Liu YF, Wu CF, Xu F, Shen ZX, Zhu YM. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA. 2009;106:3342–7.
Jácomo RH, Melo RA, Souto FR, de Mattos ER, de Oliveira CT, Fagundes EM, et al. Clinical features and outcomes of 134 Brazilians with acute promyelocytic leukemia who received ATRA and anthracyclines. Haematologica. 2007;92:1431–2.
Lehmann S, Ravn A, Carlsson L, Antunovic P, Deneberg S, Möllgård L, et al. Continuing high early death rate in acute promyelocytic leukemia: a population-based report from the Swedish Adult Acute Leukemia Registry. Leukemia. 2011;25:1128–34.
Park JH, Qiao B, Panageas KS, Schymura MJ, Jurcic JG, Rosenblat TL, et al. Early death rate in acute promyelocytic leukemia remains high despite all-trans retinoic acid. Blood. 2011;118:1248–54.
Suzuki K, Hayashi T, Nishioka J, Kosaka Y, Zushi M, Honda G, et al. A domain composed of epidermal growth factor-like structures of human thrombomodulin is essential for thrombin binding and for protein C activation. J Biol Chem. 1989;264:4872–6.
Dittman WA, Majerus PW. Structure and function of thrombomodulin: a natural anticoagulant. Blood. 1990;75:329–36.
Dahlback B, Villoutreix BO. The anticoagulation protein C pathway. FEBS Lett. 2005;579:3310–6.
Yamakawa K, Ogura H, Fujimi S, Morikawa M, Ogawa Y, Mohri T, et al. Recombinant human soluble thrombomodulin in sepsis-induced disseminated intravascular coagulation: a multicenter propensity score analysis. Intensive Care Med. 2013;39:644–52.
Ikezoe T, Ikenoue N, Uchikawa N, Kojima S, Fukaya T, Yokoyama A. Use of recombinant human soluble thrombomodulin in the management of HELLP syndrome complicated by DIC. Thromb Res. 2010;126:e238–40.
Sakai M, Ikezoe T, Bandobashi K, Yokoyama A. Successful treatment of thrombotic thrombocytopenic purpura associated with systemic lupus erythematosus with recombinant human soluble thrombomodulin. Thromb Res. 2010;2010(126):e392–3.
Uni M, Yoshimi A, Maki H, Maeda D, Nakazaki K, Nakamura F, et al. Successful treatment with recombinant thrombomodulin for B-cell lymphoma-associated hemophagocytic syndrome complicated by disseminated intravascular coagulation. Int J Clin Exp Pathol. 2013;6:1190–4.
Sakai M, Ikezoe T, Bandobashi K, Togitani K, Yokoyama A. Successful treatment of transplantation-associated thrombotic microangiopathy with recombinant human soluble thrombomodulin. Bone Marrow Transplant. 2010;45:803–5.
Ikezoe T, Togitani K, Komatsu N, Isaka M, Yokoyama A. Successful treatment of sinusoidal obstructive syndrome after hematopoietic stem cell transplantation with recombinant human soluble thrombomodulin. Bone Marrow Transplant. 2010;45:783–5.
Ikezoe T, Takeuchi A, Taniguchi A, Togitani K, Yokoyama A. Recombinant human soluble thrombomodulin counteracts capillary leakage associated with engraftment syndrome. Bone Marrow Transplant. 2011;46:616–8.
Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115:1267–74.
Conway EM, Van de Wouwer M, Pollefeyt S, Jurk K, Van Aken H, De Vriese A, et al. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor kappaB and mitogen-activated protein kinase pathways. J Exp Med. 2002;196:565–77.
Ikezoe T, Yang J, Nishioka C, Honda G, Furihata M, Yokoyama A. Thrombomodulin protects endothelial cells from a calcineurin inhibitor-induced cytotoxicity by upregulation of extracellular signal-regulated kinase/myeloid leukemia cell-1 signaling. Arterioscler Thromb Vasc Biol. 2012;32:2259–70.
Levi M, Van Der Poll T. Thrombomodulin in sepsis. Minerva Anestesiol. 2013;79:294–8.
Hoppensteadt D, Tsuruta K, Cunanan J, Hirman J, Kaul I, Osawa Y, et al. Thrombin Generation Mediators and Markers in Sepsis-Associated Coagulopathy and Their Modulation by Recombinant Thrombomodulin. Clin Appl Thromb Hemost. In press. 2013 Jun 25.
Breccia M, Avvisati G, Latagliata R, Carmosino I, Guarini A, De Propris MS, et al. Occurrence of thrombotic events in acute promyelocytic leukemia correlates with consistent immunophenotypic and molecular features. Leukemia. 2007;21:79–83.
Montesinos PJ, de la Serna, E Vellenga, C Rayon, J Bergua, R Parody et al. Incidence and risk factors for thrombosis in patients with acute promyelocytic leukemia. Experience of the PETHEMA LPA96 and LPA99 Protocols. Blood 2006: (ASH Annual Meeting Abstracts); 108: 1503.
Falanga A, Iacoviello L, Evangelista V, Consonni R, Belotti D, D’Orazio A, et al. Loss of blast cell procoagulant activity and improvement of hemostatic variables in patients with acute promyelocytic leukemia given all-trans-retinoic acid. Blood. 1995;86:1072.
Bauer KA, Rosenberg RD. Thrombin generation in acute promyelocytic leukemia. Blood. 1984;64:791.
Mjers TJ, Rickles FR, Barb C, Cronlund M. Fibrinopeptide A in acute leukemia: Relationship of activation of blood coagulation to disease activity. Blood. 1981;57:518.
Wang J, Weiss I, Svoboda K, Kwaan HC. Thrombogenic role of cells undergoing apoptosis. Br J Haematol. 2001;115:382–91.
Falanga A, Gordon SG. Isolation and characterization of cancer procoagulant: a cysteine proteinase from malignant tissue. Biochemistry. 1985;24:5558–67.
Falanga A, Alessio MG, Donati MB, Barbui T. A new procoagulant in acute leukemia. Blood. 1988;71:870–5.
Ma G, Liu F, Lv L, Gao Y, Su Y. Increased promyelocytic-derived micro particles: a novel potential factor for coagulopathy in acute promyelocytic leukemia. Ann Hematol. 2013;92:645–52.
Boles JC, Williams JC, Hollingsworth RM, Wang JG, Glover SL, Owens AP 3rd. Increased promyelocytic-derived micro particles: a novel potential factor for coagulopathy in acute promyelocytic leukemia. Ann Hematol. 2013;92:645–52.
Dubois C, Schlageter MH, de Gentile A, Guidez F, Balitrand N, Toubert ME. Hematopoietic growth factor expression and ATRA sensitivity in acute promyelocytic blast cells. Blood. 1994;83:3264–70.
Schleef RR, Bevilacqua MP, Sawdey M, Gimbrone MA Jr, Loskutoff DJ. Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J Biol Chem. 1988;263:5797–803.
Schorer AE, Kaplan ME, Rao GH, Moldow CF. Interleukin 1 stimulates endothelial cell tissue factor production and expression by a prostaglandin-independent mechanism. Thromb Haemost. 1986;56:256–9.
Booth NA, Bennett B. Plasmin-alpha-2-antiplasmin complexes in bleeding disorders characterized by primary or secondary fibrinolysis. Br J Haematol. 1984;56:545.
Reddy VB, Kowal-Vern A, Hoppensteadt DA, Kumar A, Walenga JM, Fareed J. Global and hemostatic markers in acute myeloid leukemia. Am J Clin Pathol. 1990;94:397–403.
Speiser W, Pabinger-Fasching I, Kyrle PA, Kapiotis S, Kottas-Heldenberg A, Bettelheim P. Hemostatic and fibrinolytic parameters in patients with acute myeloid leukemia: activation of blood coagulation, fibrinolysis and unspecific proteolysis. Blut. 1990;61:298–302.
Sakata Y, Murakami T, Noro A, Mori K, Matsuda M. The specific activity of plasminogen activator inhibitor-1 in disseminated intravascular coagulation with acute promyelocytic leukemia. Blood. 1991;77:1949–57.
Tapiovaara H, Alitalo R, Stephens R, Myöhänen H, Ruutu T, Vaheri A. Abundant urokinase activity on the surface of mononuclear cells from blood and bone marrow of acute leukemia patients. Blood. 1993;82:914–9.
Nadir Y, Katz T, Sarig G, Hoffman R, Oliven A, Rowe JM. Hemostatic balance on the surface of leukemic cells: the role of tissue factor and urokinase plasminogen activator receptor. Haematologica. 2005;90:1549–56.
Hajjar KA, Menell JS. Annexin II: a novel mediator of cell surface plasmin generation. Ann N Y Acad Sci. 1997;811:337–49.
Cesarman GM, Guevara CA, Hajjar KA. An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA). II. Annexin II-mediated enhancement of t-PA-dependent plasminogen activation. J Biol Chem. 1994;269:21198–203.
Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, Hajjar KA. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med. 1999;340:994–1004.
Bajzar L, Manuel R, Nesheim ME. Purification and characterization of TAFI, a thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1995;270:14477–84.
Wang W, Boffa MB, Bajzar L, Walker JB, Nesheim ME. A study of the mechanism of inhibition of fibrinolysis by activated thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1998;273:27176–81.
Redlitz A, Nicolini FA, Malycky JL, Topol EJ, Plow EF. Inducible carboxypeptidase activity. A role in clot lysis in vivo. Circulation. 1996;93:1328–30.
Meijers JC, Oudijk EJ, Mosnier LO, Bos R, Bouma BN, Nieuwenhuis HK. Reduced activity of TAFI (thrombin-activatable fibrinolysis inhibitor) in acute promyelocytic leukaemia. Br J Haematol. 2000;108:518–23.
Falanga A, Iacoviello L, Evangelista V, Belotti D, Consonni R, D’Orazio A. Loss of blast cell procoagulant activity and improvement of hemostatic variables in patients with acute promyelocytic leukemia administered all-trans-retinoic acid. Blood. 1995;86:1072–81.
Törnebohm E, Egberg N, Sablica H, Wallin R, Lockner D, Paul C. Elastase activity in leukaemic cells and plasma in patients with acute leukaemia. Eur J Haematol. 1992;49:98–104.
Oudijk EJ, Nieuwenhuis HK, Bos R, Fijnheer R. Elastase mediated fibrinolysis in acute promyelocytic leukemia. Thromb Haemost. 2000;83:906–8.
Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113:1875–91.
Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, et al. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J Thromb Haemost. 2007;5:31–41.
Ikezoe T, Takeuchi A, Isaka M, Arakawa Y, Iwabu N, Kin T, et al. Recombinant human soluble thrombomodulin safely and effectively rescues acute promyelocytic leukemia patients from disseminated intravascular coagulation. Leuk Res. 2012;36:1398–402.
Kawano N, Kuriyama T, Yoshida S, Yamashita K, Ochiai H, Nakazaki S, et al. Clinical features and treatment outcomes of six patients with disseminated intravascular coagulation resulting from acute promyelocytic leukemia and treated with recombinant human soluble thrombomodulin at a single institution. Intern Med. 2013;52:55–62.
Shindo M, Ikuta K, Addo L, Ito S, Fujiya M, Torimoto Y, et al. Successful control of disseminated intravascular coagulation by recombinant thrombomodulin during arsenic trioxide treatment in relapsed patient with acute promyelocytic leukemia. Case Rep Hematol. 2012;2012:908196.
Suzuki K, Kusumoto H, Deyashiki Y, Nishioka J, Maruyama I, Zushi M, et al. Structure and expression of human thrombomodulin, a thrombin receptor on endothelium acting as a cofactor for protein C activation. EMBO J. 1987;6:1891–7.
Stearns DJ, Kurosawa S, Esmon CT. Microthrombomodulin. Residues 310–486 from the epidermal growth factor precursor homology domain of thrombomodulin will accelerate protein C activation. J Biol Chem. 1989;264:3352–6.
Taylor FB, Stearns-Kurosawa DJ, Kurosawa S, et al. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood. 2000;95:1680–6.
Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–2.
Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–72.
Kerschen EJ, Fernandez JA, Cooley BC, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–48.
Ikezoe T, Yang J, Nishioka C, Isaka M, Iwabu N, Sakai M, et al. Thrombomodulin enhances the antifibrinolytic and antileukemic effects of all-trans retinoic acid in acute promyelocytic leukemia cells. Exp Hematol. 2012;40:457–65.
Kizaki K, Ishii H, Horie S, Kazama M. Thrombomodulin induction by all-trans retinoic acid is independent of HL-60 cells differentiation to neutrophilic cells. Thromb Haemost. 1994;72:573–7.
Dittman WA, Nelson SC, Greer PK, Horton ET, Palomba ML, McCachren SS. Characterization of thrombomodulin expression in response to retinoic acid and identification of a retinoic acid response element in the human thrombomodulin gene. J Biol Chem. 1994;269:16925–32.
Horie S, Kizaki K, Ishii H, Kazama M. Retinoic acid stimulates expression of thrombomodulin, a cell surface anticoagulant glycoprotein, on human endothelial cells. Differences between up-regulation of thrombomodulin by retinoic acid and cyclic AMP. Biochem J. 1992;281:149–54.
Frankel SR, Eardley A, Lauwers G, Weiss M, Warrell RP. The “retinoic acid syndrome” in acute promyelocytic leukemia. Ann Intern Med. 1992;117:292–6.
de Lacerda JF, do Carmo JA, Guerra ML, Geraldes J, de Lacerda JM. Multiple thrombosis in acute promyelocytic leukaemia after tretinoin. Lancet. 1993;342:114–5.
Frankel SR, Eardley A, Heller G, Berman E, Miller WH Jr, Dmitrovsky E, et al. All-trans retinoic acid for acute promyelocytic leukemia: results of the New York study. Ann Intern Med. 1994;120:278–86.
Dubois C, Schlageter MH, de Gentile A, Balitrand N, Toubert ME, Krawice I, et al. Modulation of IL-8, IL-1 beta, and G-CSF secretion by all-trans retinoic acid in acute promyelocytic leukemia. Leukemia. 1994;8:1750–7.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Ikezoe, T. Pathogenesis of disseminated intravascular coagulation in patients with acute promyelocytic leukemia, and its treatment using recombinant human soluble thrombomodulin. Int J Hematol 100, 27–37 (2014). https://doi.org/10.1007/s12185-013-1463-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-013-1463-0