
Abstract
Myelodysplastic syndromes (MDS) and related myeloid neoplasms are a heterogeneous group of myeloid neoplasms, which frequently terminate in acute myeloid leukemia (AML). During the past decade, a number of gene mutations have been identified in MDS. However, the spectrum of these mutations overlaps largely with that in AML, complicating the understanding of MDS-specific pathogenesis that discriminates MDS from AML. Recently, several groups reported frequent mutations of multiple components of the RNA splicing machinery in MDS and related disorders. Largely specific to myelodysplastic phenotypes, these splicing factor mutations provide a potential clue to better understanding of the pathogenesis of MDS.
Similar content being viewed by others


Introduction
Myelodysplastic syndromes (MDS) and related myeloid neoplasms are a heterogeneous group of intractable myeloid malignancies, characterized by deregulated blood cell production with dysplastic cell morphology and a high propensity to acute myeloid leukemia (AML) [1 ]. Over the past 10 years, significant advances have been made in our understanding of the molecular pathogenesis of MDS and other myelodysplasias, including chronic myelomonocytic leukemia (CMML) and atypical chronic myeloid leukemia, through identification of common gene mutations found in these neoplasms [2 ]. The spectrum of gene mutations in MDS and related myeloid neoplasms includes mutations involving classical signaling molecules, such as NRAS/KRAS, PTNP11, NF1, c-CBL, JAK2, and FLT3, and those involving hematopoietic transcription factors, such as RUNX1, ETV6, and CEBPA, together with TP53 and other tumor suppressor genes. In addition, the recent discovery of frequent mutations of epigenetic regulators engaged in DNA methylation (DNMT3A, TET2, and IDH1/2) and histone modifications (ASXL1 and EZH2 and other components of Polycomb complex 2) in MDS suggested a central role of a compromised epigenetic regulation in their pathogenesis [3 ]. However, many of these mutations in MDS are observed to some extent in other myeloid neoplasms, including AML and other myelproliferative neoplasms (MPNs), indicating the presence of common mechanisms among different myeloid neoplasms [4 ]. For example, DNMT3A, TET2, IDH1/2 and ASXL1 are among most frequent targets of gene mutations in both MDS and normal karyotype AML [5–10 ]. A few gene mutations are highly specific to MDS or myelodysplasia, whereas a number of chimeric genes and other mutations, including those in MPN1 and CEBPA, are highly specific to AML [11 ].
Discovery of frequent mutations of splicing machinery in myelodysplasia
Recently, several groups have reported whole exome sequencing of MDS and other myelodysplasias, in which a common finding was frequent mutations of genes involved in RNA splicing in MDS and related myeloid neoplasms [12–15 ]. Through the whole exome sequencing of nine cases with low risk MDS, of which eight were MDS with ring sideroblasts, Papaemmanuil et al. [12] identified SF3B1 mutations in six cases and in a subsequent large-scale mutation analysis, confirmed the high frequency of SF3B1 mutations in MDS (72/354; 20 %), which were rare in other myeloid neoplasms, including AML (3/57; 5 %), CML (0/53), and MPNs (12/420, 3 %). Moreover, Yoshida et al. [13] analyzed 29 cases with different subtypes of myelodysplasia and identified mutations of multiple components of the RNA splicing machinery, including SF3B1, U2AF35, SRSF2, ZRSR2, SF3A1 and PRPF40B, in 16 cases. Affecting at least eight components of the RNA splicing machinery, mutations were found in 130 of 228 MDS (57 %), 48 of 88 CMML (55 %) and 16 of 62 secondary AML (25.8 %) cases, but relatively rare in de novo AML (10/151; 6.6 %) and MPNs (5/53; 9.4 %) [13 ]. These observations were confirmed in subsequent studies [15–27 ], although splicing factor mutations seem to be rare in pediatric myeloid neoplasms, including juvenile myelomonocytic leukemia [28, 29 ].
Pathway mutations involving the RNA splicing machinery
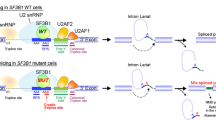
RNA splicing provides a basic cellular mechanism for expression of genetic information [30 ]. Common to all eukaryotes, this mechanism allows for generating a large diversity of protein species in the face of limited set of genes by alternative splicing [31 ]. RNA splicing is accomplished by recruitment and disengagement of multiple snRNP complexes and other protein factors to newly transcribed pre-mRNA, through which exon–intron boundaries are recognized and intronic sequences were correctly spliced out to generate mature mRNA [30 ]. RNA splicing is initiated by the recognition of 5′ splice site by a U1 snRNP complex, followed by the recruitment of a complex consisting of a U2AF35/65 heterodimer, ZRSR2 and an SR protein such as SRSF1 or 2, and other factors to recognize the 3′ splice site. Finally, a U2 snRNP complex replaces SF1 bound to the branchpoint sequence with one of its subcomponent, SF3B1, to establish a splicing A complex (Fig. 1) [ 30, 32 ]. Notably, most of the mutated splicing factors in MDS belong to the A complex, occurring largely in a mutually exclusive manner, strongly indicating that the common functional target of these mutations should be the 3′ splice site recognition (Table 1) [13 ].

After the recognition of the 5′ splice site by U1 snRNP complex, a protein complex consisting of a U2AF heterodimer, ZRSR2 and SRSF1/2 was recruited to the 3′ splice boundary, where the smaller subunit (U2AF35) of the U2AF heterodimer binds to the AG-dinucleotide, while the larger subunits recognize a polypyrimidine tract. SRSF1 and SRSF2 belong to a SR family of proteins, having one or more Serine–Arginine-rich domains, and bind to splicing enhancer sequences and also interact with other proteins through its SR domain. To the 5′ upstream of the polypyrimidine tract lies a branchpoint sequence, to which another splicing factor, SF-1, binds and together with the U2AF heterodimer and other components, participates in the establishment of the splicing E complex. Once the splicing E complex is established, a U2 snRNP complex replaces SF-1 to generate the splicing A complex. Prominently, all the major components of these splicing complex are targets of gene mutations in MDS
Presence of mutational hot spot in SF3B1, SRSF2, and U2AF35
Another conspicuous feature of splicing factor mutations is the presence of mutational hot spots in major mutational targets, including SF3B1, SRSF2 and U2AF35. In U2AF35, the mutations almost exclusively involved highly conserved two amino acid positions, S34 and Q157, within the N- and C-terminal zinc finger domains, while almost all SRSF2 mutations are missense changes at P95 or deletions involving this amino acid position [13, 17, 21, 23 ]. Less conspicuously, SF3B1 mutations were confined to 5–7 amino acid positions within the domains corresponding to exons14–15, of which ~50 % of the mutations were accounted for the K700E [12, 13 ]. No homozygous mutations have been reported for these three genes. The presence of hot spots and the absence of nonsense or frameshift changes strongly suggested that they could be associated with some gain of function rather than represented simple loss of functions. In contrast, mutations of ZRSR2 on X chromosome were distributed along the entire coding region [13 ]. About two-thirds of mutations were either nonsense or frameshift changes, causing a premature stop codon [13, 17, 23 ]. The majority of the ZRSR2 mutated cases were male, in which single mutations resulted in complete loss of functions [13 ].
Genotype–phenotype association
While compromised 3′ splice site recognition seems to be a common consequence of different splicing factor mutations, there exist strong genotype and phenotype associations for splicing factor mutations. This is most prominent for the association of SF3B1 mutations with increased ring sideroblasts. SF3B1 mutations were found in 68–82 % of refractory anemia with ring sideroblasts (RSRS) and 57–76 % of refractory cytopenia with ring sideroblasts (RCMD-RS) [12, 13, 15, 18 ]. Malcovati et al. [33 ] reported that regardless of disease type, SF3B1 mutations strongly predicted the presence of increased sideroblasts with 97.7 % positive predictive value, although it did not necessarily satisfy the criteria for RARS or RCMD-RS (i.e., 15 % of all erythroblasts). Less prominently, SRSF2 mutations were more frequently found in CMML (30.7–47 %) than in other subtypes of myeloid neoplasms [13, 21 ]. Interestingly, SF3B1 mutations, but not other splicing factor mutations, have also been reported in 5–15 % of chronic lymphocytic leukemia (CLL), especially in high-risk cases [34–38 ]. In addition, SF3B1 are mutated in several solid cancers, including breast, bladder, endometrial and other cancers, although the mutation frequencies were low [12, 39]. These genotype–phenotype associations may reflect gene-specific functions of individual mutations. For example, SF3B1 was shown to participate in Hox gene regulation through functional interaction with polycomb and trithorax genes [40 ]. SRSF2 has also been implicated in genetic stability, and its defect could lead to hypermutability [41 ].
Impact of spliceosome mutations on clinical outcome
Several reports have described the clinical impact of splicing factor mutations. However there appear to be discrepancies in this impact among different studies. Initial reports indicated a significantly better overall survival for SF3B1 mutated cases compared to unmutated cases in MDS [12, 33 ], while other studies showed no significant impact of the mutations on survival [18, 23, 27 ]. SRSF2 mutations were reported to be associated with poor prognosis in univariate analysis, but may not be an independent prognostic predictor [18, 23, 27 ]. Also, U2AF35 mutations were associated with poor prognosis or higher risk of progression to AML in univariate analysis in some series [14, 42 ] but not in others. To elucidate the exact impacts of splicing factor mutations, a well-designed control study should be required taking other common mutations also into account.
Abnormal RNA splicing caused by splicing factor mutations
The high frequency of mutations in different components of the RNA splicing machinery in MDS suggests that abnormal RNA splicing is the common consequence of these mutations, which is relevant to the pathogenesis of MDS. However, their effects on RNA splicing have been evaluated only in a very limited context. When expressed in HeLa cells, the S34F U2AF35 allele induces global defects of splicing, causing abnormal retention of intronic sequences in a wide variety of mature mRNA species [13 ]. On the other hand, the same mutation was shown to promote splicing and exon skipping of a minigene reporter in 293T cells [14 ]. These are observations in highly artificial systems; no information about abnormal splicing is available for other splicing factor mutants. To understand the role of these mutations in MDS pathogenesis, the effects of the mutant alleles on RNA splicing should be evaluated in more physiological conditions using primary hematopoietic cells. It is particularly important to identify the gene targets of possible splicing defects.
Biological impact of mutations
To date, biological consequences of splicing factor mutations have been tested only for U2AF35 mutants. Unexpectedly, S34F U2AF35 mutant-transduced HeLa cells showed severely suppressed cell growth rather than enhanced cell proliferation, accompanied by apoptosis and G2/M arrest [13 ]. The effect of the S34F and Q157P/R U2AF35 mutants was also tested in competitive repopulation assays, in which highly purified mouse hematopoietic stem cells (CD34−c-Kit+ScaI+Lin−) were transduced with each mutant and together with normal competitors, transplanted into lethally irradiated mice. In this assay, mutant transduced stem cells showed lower repopulating capacity compared to mock- or wild-type U2AF35-transduced cells, as determined by the chimerism in peripheral blood 6 weeks after transplantation [13 ], indicating that both U2AF35 mutants could disturb normal hematopoiesis. However, the lower repopulation of mutant-transduced stem cells raises a serious difficulty to our understanding of how U2AF35 mutated cells achieve clonal dominance over the remaining normal hematopoietic cells, although some oncogenes, such as oncogenic RAS, have been shown to induce apoptosis rather than transformation and promotion of cell growth depending on cell contexts [43, 44 ]. The importance of cooperation with coexisting mutations and/or the effects of abnormal bone marrow environment represent possible explanations.
Concluding remarks
Whole exome sequencing has revealed the otherwise unexpected involvement of multiple components of the RNA splicing machinery by gene mutations that characterize MDS and related myeloid neoplasms and as such, demonstrated the power of massively parallel sequencing technologies in cancer research. This discovery represents a significant advance in the field of MDS research, providing a novel clue to understanding of the pathogenesis of MDS. However, a number of critical issues remain unsolved: what is the molecular mechanism for these mutations to contribute to MDS pathogenesis, to what extent the deregulated RNA splicing could be involved in that process, what is the molecular nature of the predicted gain-of-functions, and what are the targets of these genes. Their impact on clinical parameters should also be clarified. Finally, the question of whether the recently characterized small molecular inhibitor of SF3B1 might have a therapeutic role in the treatment of myeloid neoplasms with splicing factor mutations remains to be addressed [45–47].
References
Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer. 2007;7:118–29.
Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol Off J Am Soc Clin Oncol. 2011;29:504–15.
Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612.
Levine RL, Carroll M. A common genetic mechanism in malignant bone marrow diseases. New Engl Journal Med. 2009;360:2355–7.
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. New Engl J Med. 2009;360:2289–301.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. New Engl J Med. 2010;363:2424–33.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. New Engl J Med. 2009;361:1058–66.
Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–42.
Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–15.
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. New Engl J Med. 2011;364:2496–506.
Bacher U, Schnittger S, Haferlach T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 2010;22:646–55.
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. New Engl J Med. 2011;365:1384–95.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9.
Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44:53–7.
Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia Off J Leuk Soc Am Leuk Res Fund UK. 2012;26:542–5.
Cui R, Gale RP, Xu Z, et al. Clinical importance of SF3B1 mutations in Chinese with myelodysplastic syndromes with ring sideroblasts. Leuk Res. 2012;36:1428–33.
Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C, et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012;119:3211–8.
Damm F, Thol F, Kosmider O, Kade S, Loffeld P, Dreyfus F, et al. SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implications. Leukemia Off J Leuk Soc Am Leuk Res Fund UK. 2012;26:1137–40.
Jeromin S, Haferlach T, Grossmann V, et al. High frequencies of SF3B1 and JAK2 mutations in refractory anemia with ring sideroblasts associated with marked thrombocytosis strengthen the assignment to the category of myelodysplastic/myeloproliferative neoplasms. Haematologica. 2012. doi:10.3324/haematol.2012.072538.
Lasho TL, Finke CM, Hanson CA, Jimma T, Knudson RA, Ketterling RP, et al. SF3B1 mutations in primary myelofibrosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia Off J Leuk Soc Am Leuk Res Fund UK. 2012;26:1135–7.
Meggendorfer M, Roller A, Haferlach T, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012. doi:10.1182/blood-2012-01-404863.
Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood. 2012;119:569–72.
Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012.
Visconte V, Rogers HJ, Singh J, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012. doi:10.1182/blood-2012-05-430876.
Wu SJ, Kuo YY, Hou HA, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012. doi:10.1182/blood-2012-02-412296.
Zhang SJ, Rampal R, Manshouri T, Patel J, Mensah N, Kayserian A, et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood. 2012;119:4480–5.
Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(27):3376–82.
Hirabayashi S, Flotho C, Moetter J, et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood. 2012;119(11):e96–99.
Takita J, Yoshida K, Sanada M, et al. Novel splicing-factor mutations in juvenile myelomonocytic leukemia. Leukemia Off J Leuk Soc Am Leuk Res Fund UK. 2012;26(8):1879–81.
Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–18.
Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–54.
Tronchere H, Wang J, Fu XD. A protein related to splicing factor U2AF35 that interacts with U2AF65 and SR proteins in splicing of pre-mRNA. Nature. 1997;388:397–400.
Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118:6239–46.
Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118:6904–8.
Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. New Engl J Med. 2011;365:2497–506.
Damm F, Nguyen-Khac F, Fontenay M, Bernard OA. Spliceosome and other novel mutations in chronic lymphocytic leukemia, and myeloid malignancies. Leukemia Off J Leuk Soc Am Leuk Res Fund UK. 2012;26(9):2027–31.
Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52.
Rossi D, Rasi S, Spina V, et al. Different impact of NOTCH1 and SF3B1 mutations on the risk of chronic lymphocytic leukemia transformation to Richter syndrome. Br J Haematol. 2012;158(3):426–29.
Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–60.
Isono K, Mizutani-Koseki Y, Komori T, Schmidt-Zachmann MS, Koseki H. Mammalian polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev. 2005;19:536–41.
Xiao R, Sun Y, Ding JH, Lin S, Rose DW, Rosenfeld MG, et al. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol Cell Biol. 2007;27:5393–402.
Makishima H, Visconte V, Sakaguchi H, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203–10.
Tanaka N, Ishihara M, Kitagawa M, Harada H, Kimura T, Matsuyama T, et al. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell. 1994;77:829–39.
Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37.
Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3:576–83.
Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3:570–5.
Webb TR, Joyner AS, Potter PM. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov Today. 2012 [Epub ahead of print].
Acknowledgments
This work was supported by Grants-in-Aid from the Ministry of Health, Labor and Welfare of Japan and KAKENHI (23249052, 22134006, and 21790907).
Conflict of interest
None.