
Abstract
An increasing number of cytogenetic and molecular genetic aberrations have been identified in acute myeloid leukaemia (AML), highlighting the biological heterogeneity of the disease. Moreover, the characterisation of specific molecular abnormalities provides the basis for targeted therapies, such as all trans retinoic acid (ATRA) and arsenic trioxide treatment in acute promyelocytic leukaemia or tyrosine kinase inhibitors in AML with FLT3 mutations. Several cytogenetic and molecular genetic changes have been shown to be prognostically relevant and have been acknowledged in the latest WHO classification of AML as separate entities. A detailed marker assessment at diagnosis is crucial for risk-stratification of AML patients, allowing the identification of those at high risk of relapse, who may benefit from early allogeneic stem cell transplantation. Finally, molecular markers are important for the detection of minimal residual disease after initial therapy and during long-term follow-up, which enables a more tailored treatment approach for individual AML patients.
Similar content being viewed by others
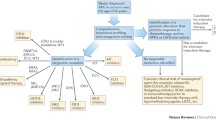


Pre-treatment prognostic factors
Cytogenetics
The leukaemic karyotype remains the most powerful predictor of prognosis in acute myeloid leukaemia (AML) and provides the framework for current risk-adapted treatment strategies adopted by different study groups (reviewed in [1]). Younger adult patients are generally categorised into 3 different risk groups based upon cytogenetics: favourable (about 20–25 %), intermediate (about 55–70 %), and adverse (about 10–20 %). While there is broad consensus about the favourable risk category, definition of intermediate and adverse risk slightly varies between the main collaborative groups for AML. Table 1 shows the revised Medical Research Council (MRC) cytogenetic risk group assignment based on the analysis of 5876 patients enrolled on the MRC AML10, 12, and 15 trials [2]. About 40–45 % of younger adult patients with AML have a normal karyotype and are generally allocated to the intermediate risk group (reviewed in [1]). Of note, adjusted risk classifications should be applied in elderly AML patients [3].
The favourable risk group includes 3 balanced translocations/inversions: t(15;17)(q22;q21)/PML-RARA in acute promyelocytic leukaemia (APL), as well as the core binding factor (CBF) leukaemias with t(8;21)(q22;q22)/RUNX1-RUNX1T1 and inv(16)(p13.1q22)/t(16;16)(p13.1;q22)/CBFB-MYH11. Patients in this group usually show a complete remission (CR) rate above 90 %, with an overall survival (OS) of 55–85 % and therefore allogeneic stem cell transplantation (SCT) in first CR is generally not recommended for these patients [4, 5].
Additional cytogenetic abnormalities do not predict for poorer outcome in AML with favourable risk cytogenetics. Indeed in CBF leukaemia with inv(16)/t(16;16), presence of additional abnormalities, particularly trisomy 22, is associated with a better outcome (reviewed in [1]).
Patients in the adverse risk group have a CR rate of only 60 % and a dismal OS of about 10–20 % [2, 6]. For these patients an allograft in first CR or experimental treatment strategies should be considered. The karyotypes −5/del(5q), −7, abn(3q), translocations involving the MLL locus at 11q23, and complex karyotypes are consistently included into the adverse risk categories of different AML study groups (reviewed in [1]).
MLL rearrangements have been generally considered to predict a poor outcome; however, different studies indicate that the prognostic impact of MLL translocations depends on the respective fusion partner. The t(6;11)(q27;q23) and t(10;11)(p12;q23), involving the AF6 and AF10 genes, respectively, were shown to confer inferior prognosis [2, 7, 8]. In contrast, the t(9;11)(p21–22;q23) involving MLLT3 and t(11;19)(q23;p13) in which the fusion partner is the ELL or ENL gene depending the breakpoint on 19p are associated with a better outcome [2, 9].
Complex karyotypes are associated with deletions of 5q, 7q and 17p (and TP53 mutations), as well as gains of 8q, 11q, and 21q [10, 11]. While various definitions have been used to categorise complex karyotypes, a recent large MRC study which investigated the impact of complexity in AML with intermediate risk aberrations suggested that 4 or more abnormalities provide the most robust and prognostically relevant cut-off [2]. A very recent study involving 824 cases conducted by the Munich Leukemia Laboratory lends further support to standardise the definition of complex karyotype based on the presence of at least 4 unrelated cytogenetic abnormalities [12].
Besides the refinement of known adverse factors, also new cytogenetic risk entities have been proposed. In a study including almost 2000 younger adults with AML, a monosomal karyotype (defined as the presence of an autosomal monosomy with at least one other autosomal monosomy or structural chromosomal abnormality; excluding CBF AML) has been associated with a particularly unfavourable prognosis and suggested to be more predictive for adverse outcome than a complex karyotype [13]. The adverse prognostic significance of a monosomal karyotype has been confirmed by other studies [2, 12, 14]. However, the vast majority of patients with a monosomal karyotype had already been defined as high risk by the revised MRC cytogenetic classification and the criterion “monosomal karyotype” alone did not reliably classify patients as high risk [2, 12].
As cytogenetic classification in AML becomes more and more complex, large prospective studies are needed to evaluate the clinical utility of known risk categories and potential new risk factors. In particular, the prognostic impact of cytogenetic aberrations of low frequency, like t(v;11)(v;q23) other than t(9;11), del(7q), del(9q), or del(20q) need to be determined. Moreover, the prognostic relevance of additional genetic abnormalities in patients with balanced translocations and other chromosomal abnormalities remains unclear and requires further evaluation.
Molecular genetics
In recent years, a magnitude of molecular prognostic factors has been identified in AML. This is particularly important for cytogenetically normal (CN)-AML patients and has substantially improved characterisation and risk-stratification of this large, heterogeneous subgroup. To date, more than 90 % of AML patients can be categorised on the basis of either cytogenetic or molecular genetic characteristics.
Thus far, 3 molecular markers [mutations in the genes encoding nucleophosmin (NPM1), Fms-like Tyrosine kinase 3 (FLT3), and CCAAT Enhancer Binding Protein alpha (C/EBPα)] have been consistently demonstrated to be of independent prognostic significance in CN-AML patients and have been widely incorporated into trial risk assignments as well as in daily clinical practice. In addition, a number of studies suggest that detection of mutations in the RUNX1 gene, partial tandem duplication of MLL (MLL-PTD), and overexpression of EVI1 confer independent adverse prognostic information and could serve to further refine risk-stratification (reviewed in [15]). The prognostic relevance of other more recently identified molecular markers, such as mutations in TET2 [16–18], IDH1/2 [19–25], DNMT3A [26–32], WT1 [33–35], KIT [36–39], ASXL1 [40–44], BCOR [45], BCORL1 [46], PHF6 [44, 47], as well as gene expression profiles (reviewed in [48]), microRNA signatures (reviewed in [49]), DNA methylation [50], or aberrant overexpression of single genes like BAALC, ERG, or MN1 (reviewed in [15]) merits further investigation.
NPM1
Heterozygous mutations of the NPM1 gene are the most frequent mutations in AML, detected in a third of cases, including approximately half with normal karyotype [51–54]. AML with mutated NPM1 has been acknowledged by the 2008 WHO classification as a provisional separate entity [55]. Mutations of NPM1 are regarded as a primary leukaemic lesion as they are stable throughout the course of the disease and are mutually exclusive with recurrent balanced translocations [56, 57]. They are associated with the presence of FLT3-ITD, IDH1/2, and DNMT3A mutations, but are rarely seen together with mutations of the CEBPA gene [54, 58].
Different NPM1 mutations have been described, all being frameshift mutations by the insertion of 4 (rarely more) bases, in the majority of cases in exon 12 [51, 59]. These mutations lead to an aberrant cytoplasmic localisation of the nucleophosmin protein and impair the protein’s nuclear shuttle function, which is important for activation and stabilisation of tumour suppressors such as p53 and ARF [60].
Patients with an isolated NPM1 mutation have a more favourable outcome, with higher CR rates, reduced risk of relapse and longer OS [52–54, 61]. However, in most studies this favourable prognosis was only seen in patients with wildtype FLT3. Moreover, the prognostic impact of NPM1 might depend on the presence of additional IDH1/2 mutations [44]. A favourable outcome associated with NPM1 was also observed in elderly AML patients [62] as well as in relapsed AML cases [63].
FLT3
About 20–27 % of AML arising in younger adults and 30–50 % of those with normal karyotype harbour in-frame internal tandem duplications (ITD) of the class III receptor tyrosine kinase FLT3 [64–66]. FLT3 is expressed on normal haematopoietic progenitor cells and is important for cell differentiation, proliferation, and survival [67]. Most FLT3-ITD mutations occur in exons 14 and 15, affecting the juxtamembrane domain which regulates the kinase activity of the receptor [68]. The duplications, which differ in length and insertion sites, lead to constitutive activation of the receptor and promote proliferation of leukaemic cells [69]. CN-AML patients with FLT3-ITD show a significantly inferior outcome compared to patients without the mutation [65, 66, 70, 71]. This is predominantly due to a higher relapse rate in cases with FLT3-ITD, whereas the initial response to chemotherapy is similar. The prognosis is particularly poor for patients with a high FLT3-ITD allelic ratio which may stem from homozygous mutations resulting from acquired uniparental disomy, as well as for patients harbouring the ITD integration site in the beta1-sheet of the tyrosine kinase domain (TKD)-1 [72] and for cases with coexisting mutations of WT1 [34, 73].
The prognostic significance of another, less frequent class of FLT3 mutations involving amino acid substitutions in the TKD (mostly at codons D835 and I836) is still unclear and might depend on the presence of other mutations [74–77]. Both ITD and TKD point mutations of FLT3 are associated with a high white blood cell count and increased LDH levels at diagnosis [71]. FLT3 mutations are more frequent in PML-RARA+ APL and are associated with the clinically aggressive microgranular variant subtype [78–80].
CEBPA
The gene CEBPA is mutated in approximately 5–10 % of AML, mostly CN-AML cases [54, 81]. In addition, germline CEBPA mutations have been reported in familial AML [82]. C/EBPα is a key transcription factor involved in myelopoiesis [83]. Mutations cluster in the amino- and carboxy-terminal regions, with the former leading to expression of a truncated isoform of C/EBPα (p30) and loss of the full length protein (p42) [81]. Carboxy-terminal mutations affect regions involved in mediating dimerisation and DNA-binding. Interestingly, in the majority of patients with CEBPA mutations, both alleles are involved, combining an upstream mutation in one allele with a downstream mutation in the other. Significant insights into the biology of CEBPA mutations have been provided by murine models, which have shown how loss of p42 expression (mimicking biallelic amino-terminal CEBPA mutations) or compound heterozygous mutations affecting amino- and carboxy-terminal regions affect haematopoiesis and give rise to AML [84]. While early studies reported that CEBPA mutation predicts a relatively favourable outcome in AML, it has subsequently been shown that this effect is accounted for by the subset of patients with biallelic mutations, especially those who lack FLT3-ITD [58, 85]. Similar to NPM1, CEBPA-mutated AML has been recognised as a provisional entity in the 2008 WHO classification [55].
Different studies have investigated outcome of patients with NPM1, FLT3, and CEBPA mutations and their mutual influence on prognosis [52–54, 58, 85]. Importantly, AML with wildtype FLT3 and mutated NPM1 or biallelic CEBPA mutations is associated with a relatively favourable prognosis similar to CBF leukaemia (see Fig. 1) and therefore patients with these molecularly defined subsets are not considered candidates for an allograft in first CR [54, 58, 86]. The genotypes “mutated NPM1 without FLT3-ITD” and “mutated CEBPA” in CN-AML have been incorporated into the favourable risk category of the recent European LeukemiaNet classification [87].

Outcome of younger adults with AML according to cytogenetic and molecular abnormalities. Overall survival for younger adults treated in the MRC AML10 and AML12 trials screened for NPM1, CEBPA and FLT3-ITD mutations [58, 61] for whom cytogenetic data were available. Cases were classified in hierarchical fashion with t(15;17)(q22;q21), t(8;21)(q22;q22), inv(16)(p13q22)/t(16;16)(p13;q22), t(9;11)(p21–22;q23), t(6;9)(p23;q34) and inv(3)(q21q26)/t(3;3)(q21;q26) at the top of the hierarchy, then CEBPA biallelic mutations, NPM1 mut/FLT3-ITDneg, NPM1 wt/FLT3-ITDpos, other intermediate and other adverse cytogenetic abnormalities
Epigenetic modifiers
TET2 Mutations of the TET2 gene have been found in various myeloid malignancies and are detected in 10–20 % of AML [17, 18, 88, 89]. TET2 mutations are very heterogeneous and spread throughout the entire coding sequence [17, 18, 88]. They are loss-of-function mutations, in most cases nonsense and frameshift mutations leading to truncated translation. The detection of mutations in both TET2 copies and loss of heterozygosity in different myeloid neoplasms suggest a role of TET2 as a tumour suppressor gene [16, 90, 91]. In vitro and in vivo studies have demonstrated a role for TET2 in myeloid differentiation and self-renewal of stem and progenitor cells [89, 92, 93]; however, the precise mechanisms and downstream effects of TET2 are yet unknown. The TET2 protein is an enzyme catalysing the conversion of 5-methylcytosine to 5-hydroxymethylcytosine which results in demethylation of DNA [94] and TET2 mutations have been shown to impair this enzymatic function [92].
The prognostic relevance of TET2 remains controversial—some studies suggest an adverse impact of TET2 mutations on outcome in certain AML subgroups [17, 18]; in other studies no prognostic significance of TET2 was found [95].
IDH Mutations of the gene encoding the isocitrate dehydrogenase IDH1 in AML were discovered by whole genome sequencing [19] and are detected in 8–16 % of cases with normal karyotype [19, 21]. Another 12–15 % (which do not harbour an IDH1 mutation) show mutations in IDH2 [20, 96], the mitochondrial homolog of IDH1. Both enzymes convert isocitrate to α-ketoglutarate; however, the mutated proteins exhibit a gain-of-function leading to aberrant accumulation of the oncometabolite 2-hydroxyglutarate (2-HG) [96, 97]. There is a functional overlap between IDH1/2 and TET2 and the two classes of mutations are mutually exclusive [17, 18, 89]. Patients with IDH1/2 and TET2 mutations show a similar epigenetic signature and global DNA hypermethylation [89]. Moreover, it was demonstrated that 2-HG which is generated as a result of mutations in IDH1/2 inhibits TET2 function [89].
Like for TET2, the impact of IDH mutations on survival of AML patients is not clear yet. Some studies have observed no difference in outcome with respect to the IDH mutation status [20, 21, 25], others have demonstrated a poor prognostic impact in certain AML subgroups [22–24]. Recent studies suggest that the impact of IDH2 on prognosis depends on the mutation site, with the IDH2 R140 mutation being an independent favourable prognostic factor in AML patients [44, 98, 99].
Apart from potential prognostic implications of IDH and TET2, the finding of alterations in enzymatic activities and epigenetic changes in a large proportion of AML patients might lead to the development of new targeted therapies for this patient group.
DNMT3A Mutations of DNMT3A occur in about 14–18 % of AML, including 20–35 % with normal karyotype and are associated with a poorer prognosis [26–32]. DNMT3A mutations are associated with the presence of NPM1, FLT3 and IDH mutations [26, 30]. Several different loss-of-function mutations have been found in all exons of DNMT3A, most frequently a missense point mutation at amino acid R882. DNMT3A is a methyltransferase that converts cytosine to 5-methylcytosine and generates de novo DNA methylation. Aberrant methylation patterns have been implicated in tumourigenesis and tumour progression [100]. The functional consequences of DNMT3A mutations in leukaemia are barely understood. It is suggested that mutations result in a decreased methyltransferase activity of DNMT3A by a dominant-negative mechanism [26, 101]. A recent in vivo study revealed that Dnmt3a-null haematopoietic stem cells have an increased self-renewal capacity and lose their differentiation potential, which was accompanied by hypomethylation of several genes implicated in leukaemogenesis [102]. However, also marked focal hypermethylation was found in the absence of Dnmt3a and it thus remains unclear which biological function of Dnmt3a contributes to the Dnmt3a-null phenotype. Interestingly, knockout of Dnmt3a alone was not sufficient to initiate leukaemia [102].
Minimal residual disease (MRD) assessment
Response to initial chemotherapy is a strong prognostic factor in AML (reviewed in [15]). Using cytogenetic analysis including FISH, polymerase chain reaction (PCR) or flow cytometry, the depth of remission and risk of relapse can be determined much more accurately than with conventional cytological methods. With PCR- and flow cytometry-based techniques, the presence of sub-microscopic levels of residual leukaemia can be detected with a sensitivity of up to 1 AML cell in 104–106 bone marrow cells (reviewed in [103]).
MRD assessment has been applied in 2 different ways: first, at early timepoints to determine the depth of the initial therapy response to better risk-stratify patients and guide postremission therapy; and second, sequential monitoring utilising leukaemia-specific markers to identify an impending relapse, enabling early treatment intervention to prevent progression of the disease.
Real-time quantitative PCR
The method with highest sensitivity for MRD analysis is real-time quantitative PCR (RT-qPCR) for AML-specific target fusion genes or mutations. So far, assays have been successfully performed for PML-RARA, RUNX1-RUNX1T1, CBFB-MYH11, MLLT3-MLL, DEK-CAN, and NPM1 mutations (reviewed in [103]). Given the increasing number of molecular markers identified in AML, at least half of patients harbour a leukaemia-specific target that can potentially be used for PCR-based MRD monitoring. However, not every marker is equally suitable for MRD assessment. For example, the use of FLT3-ITD seems to be limited, since mutational status may differ between leukaemic subclones such that the FLT3-ITD associated with the predominant clone at diagnosis may not be detectable at the time of disease relapse [104–106]. CEBPA mutations are difficult to track due to their heterogeneity and MRD detection of CEBPA has only been performed in one study including 4 patients so far [107]. Another restriction for MRD utility is the background amplification level of markers in the normal bone marrow, which has been a relevant drawback for monitoring of MLL-PTD [108].
The use of MRD assessment to guide therapy is most advanced in APL, where a standardised RT-qPCR assay has been developed and MRD monitoring is recommended by international treatment guidelines [109, 110]. Longitudinal monitoring of PML-RARA has been shown to be a strong prognostic factor for clinical relapse [110, 111]. Moreover, MRD-guided pre-emptive therapy reduced the rate of overt clinical relapse and was associated with improved survival, particularly in patients with high risk disease [111–113]. In CBF leukaemias, use of RT-qPCR to assess kinetics of disease response predicts subsequent risk of relapse [114–116]. Although, the clinical utility of sequential MRD monitoring in RUNX1-RUNX1T1-associated leukaemias has been questioned due to early reports involving qualitative PCR assays in which fusion transcripts could be detected in patients in long-term remission, subsequent studies using RT-qPCR have shown that relapse of CBF leukaemia is predicted by a rising transcript level.
Several studies have investigated NPM1 mutations as targets for MRD detection using DNA- or RNA-based quantitative PCR assays [117–121]. It has been shown that NPM1-specific monitoring independently predicted the risk of relapse [119]. Moreover, tracking of NPM1 has been successfully applied in the post-transplantation setting and predicted relapse earlier than assessment of chimaerism [120].
Besides leukaemia-specific mutations or fusion genes, aberrantly overexpressed genes have been considered as potential MRD markers, provided that they are at least 1,000-fold higher expressed than in normal cells and not modulated by regeneration after chemotherapy (reviewed in [103]). The main focus in this regard has been on WT1, which is overexpressed in about 70 % of AML cases. Early WT1 monitoring has been shown to independently predict the risk of relapse within cytogenetic risk groups; however, high background expression of WT1 in normal haematopoietic cells limits the sensitivity of WT1 assays and makes it inappropriate for long-term follow-up [122].
Flow cytometry
For patients who have no suitable molecular MRD marker, monitoring by flow cytometry may be applicable. This technique is based upon the identification of a leukaemia-associated aberrant immunophenotype (LAIP) at diagnosis that is uncommon in normal haematopoietic cells and in regenerating bone marrow. In the majority of cases, LAIPs can be identified in the blast population, which comprise cross-lineage expression of lymphoid antigens, asynchronous antigen expression, and up- or downregulation of antigens (reviewed in [103]). For MRD detection, either a fixed panel of antibodies or patient-specific antibodies may be used. As the leukaemic immunophenotype frequently changes in AML at relapse, it is recommended to use antibody panels which detect all LAIPs present at diagnosis in order to ensure the stable detection of at least one LAIP during the course of the disease (reviewed in [123]). These antigen shifts may be caused by the selection of leukaemic subclones during chemotherapy which were not detectable at diagnosis or to a lesser extent by clonal evolution of the blast population. A major problem of using flow cytometry for MRD detection is the large assay variability between different laboratories. Nevertheless, several studies have demonstrated the independent prognostic significance of MRD assessment with flow cytometry after induction and consolidation therapy [124–126]. Thus far, sensitivity of MRD detection using flow cytometry is significantly lower than that of RT-qPCR, which limits the method to early MRD assessment. However, this will be probably overcome with the use of advanced multicolour technologies in the future.
Both for PCR- and flow cytometry-based MRD approaches, large prospective trials are needed to evaluate the clinical utility of MRD in non-APL AML. In this regard, it is particularly important to further standardise the different MRD assays and to equalise time points and cut-offs for MRD detection. Most likely, MRD assessment will provide a useful tool for risk-stratification during therapy and follow-up and may improve outcome for AML patients.
Concluding remarks
The identification of molecular markers in AML has greatly improved our understanding of disease pathogenesis and facilitated the discrimination of biologically and clinically distinct subgroups. Cytogenetic analysis and molecular screening for NPM1, FLT3 and CEBPA mutations have been widely adopted into the routine diagnostic work-up of AML, informing treatment decisions including the application of allogeneic SCT in first CR. The increasing number of molecular markers identified as a result of recent high throughput sequencing initiatives has presented a considerable challenge to establish the prognostic significance of particular constellations of mutations which may be relatively uncommon, demanding analysis of very large numbers of homogeneously treated patients. Nevertheless, it seems likely that these technologies will lead to the identification of new molecular markers with independent prognostic impact, which will improve prediction of an individual patient’s response to therapy. It is anticipated that further subgroups of patients who may benefit from particular (targeted) therapeutic approaches will be identified. In addition, technical improvements in flow cytometry with an increased number of available colours and the capacity to identify leukaemic stem cell populations, coupled with the development of an expanding range of RT-qPCR assays for leukaemia-specific molecular targets, make it highly plausible that longitudinal tracking of MRD can be applied in virtually every case of AML in the near future. These approaches could provide powerful tools to individually tailor a patient’s treatment throughout the entire course of the disease. This prospect will be extremely pertinent to the design of the next wave of large scale clinical trials which will need to establish the most informative molecular markers and determine whether the use of MRD monitoring to guide therapy is clinically useful, leading to meaningful improvements in patient survival.
References
Grimwade D, Mrozek K. Diagnostic and prognostic value of cytogenetics in acute myeloid leukemia. Hematol Oncol Clin North Am. 2011;25:1135–61. (vii).
Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–65.
Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001;98:1312–20.
Burnett AK, Wheatley K, Goldstone AH, Stevens RF, Hann IM, Rees JH, et al. The value of allogeneic bone marrow transplant in patients with acute myeloid leukaemia at differing risk of relapse: results of the UK MRC AML 10 trial. Br J Haematol. 2002;118:385–400.
Cornelissen JJ, van Putten WL, Verdonck LF, Theobald M, Jacky E, Daenen SM, et al. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle-aged adults: benefits for whom? Blood. 2007;109:3658–66.
Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–83.
Blum W, Mrozek K, Ruppert AS, Carroll AJ, Rao KW, Pettenati MJ, et al. Adult de novo acute myeloid leukemia with t(6;11)(q27;q23): results from Cancer and Leukemia Group B Study 8461 and review of the literature. Cancer. 2004;101:1420–7.
Krauter J, Wagner K, Schafer I, Marschalek R, Meyer C, Heil G, et al. Prognostic factors in adult patients up to 60 years old with acute myeloid leukemia and translocations of chromosome band 11q23: individual patient data-based meta-analysis of the German Acute Myeloid Leukemia Intergroup. J Clin Oncol. 2009;27:3000–6.
von Neuhoff C, Reinhardt D, Sander A, Zimmermann M, Bradtke J, Betts DR, et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol. 2010;28:2682–9.
Rucker FG, Bullinger L, Schwaenen C, Lipka DB, Wessendorf S, Frohling S, et al. Disclosure of candidate genes in acute myeloid leukemia with complex karyotypes using microarray-based molecular characterization. J Clin Oncol. 2006;24:3887–94.
Mrozek K. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol. 2008;35:365–77.
Haferlach C, Alpermann T, Schnittger S, Kern W, Chromik J, Schmid C, et al. Prognostic value of monosomal karyotype in comparison to complex aberrant karyotype in acute myeloid leukemia: a study on 824 cases with aberrant karyotype. Blood. 2012;119:2122–5.
Breems DA, Van Putten WL, De Greef GE, Van Zelderen-Bhola SL, Gerssen-Schoorl KB, Mellink CH, et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;26:4791–7.
Kayser S, Zucknick M, Dohner K, Krauter J, Kohne CH, Horst HA, et al. Monosomal karyotype in adult acute myeloid leukemia: prognostic impact and outcome after different treatment strategies. Blood. 2012;119:551–8.
Smith ML, Hills RK, Grimwade D. Independent prognostic variables in acute myeloid leukaemia. Blood Rev. 2011;25:39–51.
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301.
Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–81.
Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–10.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66.
Thol F, Damm F, Wagner K, Gohring G, Schlegelberger B, Hoelzer D, et al. Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. 2010;116:614–6.
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010;115:2749–54.
Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–55.
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–43.
Schnittger S, Haferlach C, Ulke M, Alpermann T, Kern W, Haferlach T. IDH1 mutations are detected in 6.6 % of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood. 2010;116:5486–96.
Ravandi F, Patel K, Luthra R, Faderl S, Konopleva M, Kadia T, et al. Prognostic significance of alterations in IDH enzyme isoforms in patients with AML treated with high-dose cytarabine and idarubicin. Cancer 2012;118:2665–73.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33.
Thol F, Damm F, Ludeking A, Winschel C, Wagner K, Morgan M, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol. 2011;29:2889–96.
Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118:5593–603.
Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119:559–68.
Renneville A, Boissel N, Nibourel O, Berthon C, Helevaut N, Gardin C, et al. Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: a study by the Acute Leukemia French Association. Leukemia 2012;26:1247–54.
Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrozek K, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol 2012;30:742–50.
Markova J, Michkova P, Burckova K, Brezinova J, Michalova K, Dohnalova A, et al. Prognostic impact of DNMT3A mutations in patients with intermediate cytogenetic risk profile acute myeloid leukemia. Eur J Haematol. 2012;88:128–35.
King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms’ tumor gene WT1 in leukemias. Blood. 1996;87:2171–9.
Virappane P, Gale R, Hills R, Kakkas I, Summers K, Stevens J, et al. Mutation of the Wilms’ tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol. 2008;26:5429–35.
Paschka P, Marcucci G, Ruppert AS, Whitman SP, Mrozek K, Maharry K, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2008;26:4595–602.
Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia. 2006;20:965–70.
Paschka P, Marcucci G, Ruppert AS, Mrozek K, Chen H, Kittles RA, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24:3904–11.
Cairoli R, Beghini A, Grillo G, Nadali G, Elice F, Ripamonti CB, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood. 2006;107:3463–8.
Schnittger S, Kohl TM, Haferlach T, Kern W, Hiddemann W, Spiekermann K, et al. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood. 2006;107:1791–9.
Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145:788–800.
Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116:4086–94.
Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrozek K, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118:6920–9.
Pratcorona M, Abbas S, Sanders MA, Koenders JE, Kavelaars FG, Erpelinck-Verschueren CA, et al. Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica. 2012;97:388–92.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89.
Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118:6153–63.
Li M, Collins R, Jiao Y, Ouillette P, Bixby D, Erba H, et al. Somatic mutations in the transcriptional corepressor gene BCORL1 in adult acute myelogenous leukemia. Blood. 2011;118:5914–7.
Van Vlierberghe P, Patel J, Abdel-Wahab O, Lobry C, Hedvat CV, Balbin M, et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia. 2011;25:130–4.
Mano H. Stratification of acute myeloid leukemia based on gene expression profiles. Int J Hematol. 2004;80:389–94.
Marcucci G, Mrozek K, Radmacher MD, Garzon R, Bloomfield CD. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood. 2011;117:1121–9.
Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–66.
Dohner K, Schlenk RF, Habdank M, Scholl C, Rucker FG, Corbacioglu A, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–6.
Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107:4011–20.
Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2008.
Haferlach C, Mecucci C, Schnittger S, Kohlmann A, Mancini M, Cuneo A, et al. AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biologic, pathologic, immunophenotypic, and prognostic features. Blood. 2009;114:3024–32.
Falini B, Mecucci C, Saglio G, Lo Coco F, Diverio D, Brown P, et al. NPM1 mutations and cytoplasmic nucleophosmin are mutually exclusive of recurrent genetic abnormalities: a comparative analysis of 2562 patients with acute myeloid leukemia. Haematologica. 2008;93:439–42.
Green CL, Koo KK, Hills RK, Burnett AK, Linch DC, Gale RE. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J Clin Oncol. 2010;28:2739–47.
Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc + AML): biologic and clinical features. Blood. 2007;109:874–85.
Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493–505.
Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–84.
Becker H, Marcucci G, Maharry K, Radmacher MD, Mrozek K, Margeson D, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:596–604.
Chevallier P, Prebet T, Pigneux A, Hunault M, Delaunay J, Perry F, et al. Influence of NPM1 and FLT3-ITD status on outcome in relapsed/refractory AML patients receiving salvage therapy including gemtuzumab ozogamicin. Leukemia. 2010;24:467–9.
Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8.
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–9.
Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35.
Kiyoi H, Naoe T. FLT3 in human hematologic malignancies. Leuk Lymphoma. 2002;43:1541–7.
Kiyoi H, Towatari M, Yokota S, Hamaguchi M, Ohno R, Saito H, et al. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia. 1998;12:1333–7.
Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. 2002;21:2555–63.
Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a Cancer and Leukemia Group B study. Cancer Res. 2001;61:7233–9.
Frohling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–80.
Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114:2386–92.
Gaidzik VI, Schlenk RF, Moschny S, Becker A, Bullinger L, Corbacioglu A, et al. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German–Austrian AML Study Group. Blood. 2009;113:4505–11.
Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia. 2005;19:1345–9.
Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. 2007;110:1262–70.
Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters—an analysis of 3082 patients. Blood. 2008;111:2527–37.
Whitman SP, Ruppert AS, Radmacher MD, Mrozek K, Paschka P, Langer C, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. 2008;111:1552–9.
Kiyoi H, Naoe T, Yokota S, Nakao M, Minami S, Kuriyama K, et al. Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia Study Group of the Ministry of Health and Welfare (Kohseisho). Leukemia. 1997;11:1447–52.
Noguera NI, Breccia M, Divona M, Diverio D, Costa V, De Santis S, et al. Alterations of the FLT3 gene in acute promyelocytic leukemia: association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia. 2002;16:2185–9.
Gale RE, Hills R, Pizzey AR, Kottaridis PD, Swirsky D, Gilkes AF, et al. Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood. 2005;106:3768–76.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–70.
Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351:2403–7.
Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–14.
Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13:299–310.
Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–91.
Barjesteh van Waalwijk van Doorn-Khosrovani S, Erpelinck C, Meijer J, van Oosterhoud S, van Putten WL, Valk PJ, et al. Biallelic mutations in the CEBPA gene and low CEBPA expression levels as prognostic markers in intermediate-risk AML. Hematol J. 2003;4:31–40.
Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74.
Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–7.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–42.
Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113:6403–10.
Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3.
Gaidzik VI, Paschka P, Späth D, Habdank M, Köhne CH, Germing U, et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML Study Group. J Clin Oncol. 2012;30:1350–7.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34.
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–44.
Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, et al. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood. 2011;118:409–12.
Boissel N, Nibourel O, Renneville A, Huchette P, Dombret H, Preudhomme C. Differential prognosis impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. 2011;117:3696–7.
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98.
Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–15.
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31.
Freeman SD, Jovanovic JV, Grimwade D. Development of minimal residual disease-directed therapy in acute myeloid leukemia. Semin Oncol. 2008;35:388–400.
Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100:2393–8.
Palmisano M, Grafone T, Ottaviani E, Testoni N, Baccarani M, Martinelli G. NPM1 mutations are more stable than FLT3 mutations during the course of disease in patients with acute myeloid leukemia. Haematologica. 2007;92:1268–9.
Shih LY, Huang CF, Wu JH, Lin TL, Dunn P, Wang PN, et al. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002;100:2387–92.
Smith LL, Pearce D, Smith ML, Jenner M, Lister TA, Bonnet D, et al. Development of a quantitative real-time polymerase chain reaction method for monitoring CEBPA mutations in normal karyotype acute myeloid leukaemia. Br J Haematol. 2006;133:103–5.
Weisser M, Kern W, Schoch C, Hiddemann W, Haferlach T, Schnittger S. Risk assessment by monitoring expression levels of partial tandem duplications in the MLL gene in acute myeloid leukemia during therapy. Haematologica. 2005;90:881–9.
Milligan DW, Grimwade D, Cullis JO, Bond L, Swirsky D, Craddock C, et al. Guidelines on the management of acute myeloid leukaemia in adults. Br J Haematol. 2006;135:450–74.
Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113:1875–91.
Grimwade D, Jovanovic JV, Hills RK, Nugent EA, Patel Y, Flora R, et al. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J Clin Oncol. 2009;27:3650–8.
Lo Coco F, Diverio D, Avvisati G, Petti MC, Meloni G, Pogliani EM, et al. Therapy of molecular relapse in acute promyelocytic leukemia. Blood. 1999;94:2225–9.
Esteve J, Escoda L, Martin G, Rubio V, Diaz-Mediavilla J, Gonzalez M, et al. Outcome of patients with acute promyelocytic leukemia failing to front-line treatment with all-trans retinoic acid and anthracycline-based chemotherapy (PETHEMA protocols LPA96 and LPA99): benefit of an early intervention. Leukemia. 2007;21:446–52.
Krauter J, Gorlich K, Ottmann O, Lubbert M, Dohner H, Heit W, et al. Prognostic value of minimal residual disease quantification by real-time reverse transcriptase polymerase chain reaction in patients with core binding factor leukemias. J Clin Oncol. 2003;21:4413–22.
Perea G, Lasa A, Aventin A, Domingo A, Villamor N. Queipo de Llano MP, et al. Prognostic value of minimal residual disease (MRD) in acute myeloid leukemia (AML) with favorable cytogenetics [t(8;21) and inv(16)]. Leukemia. 2006;20:87–94.
Corbacioglu A, Scholl C, Schlenk RF, Eiwen K, Du J, Bullinger L, et al. Prognostic impact of minimal residual disease in CBFB-MYH11-positive acute myeloid leukemia. J Clin Oncol. 2010;28:3724–9.
Gorello P, Cazzaniga G, Alberti F, Dell’Oro MG, Gottardi E, Specchia G, et al. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia. 2006;20:1103–8.
Chou WC, Tang JL, Wu SJ, Tsay W, Yao M, Huang SY, et al. Clinical implications of minimal residual disease monitoring by quantitative polymerase chain reaction in acute myeloid leukemia patients bearing nucleophosmin (NPM1) mutations. Leukemia. 2007;21:998–1004.
Schnittger S, Kern W, Tschulik C, Weiss T, Dicker F, Falini B, et al. Minimal residual disease levels assessed by NPM1 mutation-specific RQ-PCR provide important prognostic information in AML. Blood. 2009;114:2220–31.
Bacher U, Badbaran A, Fehse B, Zabelina T, Zander AR, Kroger N. Quantitative monitoring of NPM1 mutations provides a valid minimal residual disease parameter following allogeneic stem cell transplantation. Exp Hematol. 2009;37:135–42.
Krönke J, Schlenk RF, Jensen KO, Tschürtz F, Corbacioglu A, Gaidzik VI, et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German–Austrian acute myeloid leukemia study group. J Clin Oncol. 2011;29:2709–16.
Cilloni D, Renneville A, Hermitte F, Hills RK, Daly S, Jovanovic JV, et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: a European LeukemiaNet study. J Clin Oncol. 2009;27:5195–201.
Kern W, Bacher U, Haferlach C, Schnittger S, Haferlach T. The role of multiparameter flow cytometry for disease monitoring in AML. Best Pract Res Clin Haematol. 2010;23:379–90.
Venditti A, Buccisano F, Del Poeta G, Maurillo L, Tamburini A, Cox C, et al. Level of minimal residual disease after consolidation therapy predicts outcome in acute myeloid leukemia. Blood. 2000;96:3948–52.
San Miguel JF, Vidriales MB, Lopez-Berges C, Diaz-Mediavilla J, Gutierrez N, Canizo C, et al. Early immunophenotypical evaluation of minimal residual disease in acute myeloid leukemia identifies different patient risk groups and may contribute to postinduction treatment stratification. Blood. 2001;98:1746–51.
Maurillo L, Buccisano F, Del Principe MI, Del Poeta G, Spagnoli A, Panetta P, et al. Toward optimization of postremission therapy for residual disease-positive patients with acute myeloid leukemia. J Clin Oncol. 2008;26:4944–51.
Acknowledgments
DG gratefully acknowledges the National Institute for Health Research (NIHR) for support for molecular diagnostics and assessment of minimal residual disease in the UK NCRI AML17 trial. This paper presents independent research funded by the National Institute for Health Research (NIHR) under its Programme Grants for Applied Research Programme (Grant Reference Number RP- PG-0108-10093). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. In addition, DG gratefully acknowledges research funding from Leukaemia & Lymphoma Research of Great Britain, the Guy’s and St. Thomas’ Charity and the MRD Workpackage (WP12) of the European LeukemiaNet. AK acknowledges financial support from the Deutsche Krebshilfe. The authors also thank Dr Robert Hills for the data analyses summarised in Fig. 1.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Kühnl, A., Grimwade, D. Molecular markers in acute myeloid leukaemia. Int J Hematol 96, 153–163 (2012). https://doi.org/10.1007/s12185-012-1123-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-012-1123-9