
Abstract
Sorption isotherms of hydrocarbon and carbon dioxide (CO2) provide crucial information for designing processes to sequester CO2 and recover natural gas from unmineable coal beds. Methane (CH4), ethane (C2H6), and CO2 adsorption isotherms on dry coal and the temperature effect on their maximum sorption capacity have been studied by performing combined Monte Carlo (MC) and molecular dynamics (MD) simulations at temperatures of 308 and 370 K (35 and 97 °C) and at pressures up to 10 MPa. Simulation results demonstrate that absolute sorption (expressed as a mass basis) divided by bulk gas density has negligible temperature effect on CH4, C2H6, and CO2 sorption on dry coal when pressure is over 6 MPa. CO2 is more closely packed due to stronger interaction with coal and the stronger interaction between CO2 molecules compared, respectively, with the interactions between hydrocarbons and coal and between hydrocarbons. The results of this work suggest that the “a” constant (proportional to T 2 c /P c ) in the Peng–Robinson equation of state is an important factor affecting the sorption behavior of hydrocarbons. CO2 injection pressures of lower than 8 MPa may be desirable for CH4 recovery and CO2 sequestration. This study provides a quantitative understanding of the effects of temperature on coal sorption capacity for CH4, C2H6, and CO2 from a microscopic perspective.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Enhanced coalbed methane (ECBM) is increasingly important unconventional gas. Carbon dioxide (CO2) injection in coal seams can replace hydrocarbons and release renewable energy. Understanding the mechanism of CH4, C2H6, and CO2 sorption on coal is a key factor for CO2 storage and hydrocarbons recovery. Sorption of gases in coal has been studied for decades (Bae and Bhatia 2006; Billemont et al. 2011, 2013; Brochard et al. 2012a, 2012b; Busch and Gensterblum 2011; Busch et al. 2003, 2004; Day et al. 2008; Fitzgerald et al. 2005; Gensterblum et al. 2014; Goodman et al. 2007; Krooss et al. 2002; Li et al. 2010; Ottiger et al. 2008; Pini et al. 2009, 2010). Theoretical models have been developed and improved to study gas sorption on coal by several authors (Connell et al. 2010; Lu and Connell 2007; Lu et al. 2008; Pan and Connell 2007, 2009, 2012; Sakurovs et al. 2007; Vandamme et al. 2010). They have applied different approaches to describe how sorption capacity, sorption rate, gas diffusion, and permeability are affected. Several groups reported that the sorption of gas on coals at a given pressure increases with decreasing temperature (Azmi et al. 2006; Menon 1968; White et al. 2005; Zhou et al. 2001), but the temperature effect on the maximum sorption capacity was controversial (Sakurovs et al. 2010), nor was the temperature effect quantified. Major challenges for ECBM lie in the fact that the density of the adsorbed phase within varying pores is unknown and fundamental understanding of the sorption mechanism is insufficient. In this study, we simulate sorption of CH4, C2H6, and CO2 on dry intermediate rank bituminous coal at 308 and 370 K up to a pressure of 10 MPa. The temperature effect on sorption capacity of CH4, C2H6, and CO2 and the interactions between adsorbate and coal and between adsorbate are investigated in an attempt to provide an insight into the sorption mechanism of coal for those gases, as well as to demonstrate the potential use of this simulation method for ECBM.
The remainder of this article is structured as follows. In Sect. 2, we explain the molecular models selected for coal, CH4, C2H6, and CO2, the methods used, and the implementation of the simulations. In Sect. 3, we present and discuss the results of the molecular simulations. Finally, we summarize our analysis and draw conclusions in Sect. 4.
2 Simulation details
Molecular simulation allows one to describe the interactions between the adsorbate species and between the adsorbate and coal matrix in full detail, without predefined density profiles or sorption patterns. Its advantage lies in its ability to predict micro sorption and to reveal the mechanism of gas adsorption.
2.1 Simulation setup
Coal is characterized by two distinct porosity systems: micropores or matrix and macropores or cleats. The matrix is storage medium where coal seam gas (primarily CH4 and CO2) is mainly stored by sorption and moves by molecular diffusion. The cleats constitute a natural fracture network and provide permeability and connectivity to the reservoir but very limited storage volume as free gas.
We simulate gas sorption in the coal matrix. The system studied consists of coal and pure components of CO2, CH4, and C2H6. In this study, we focus on a model representation of a bituminous coal (Spiro and Kosky 1982; Tambach et al. 2009). A building block of 191 atoms, C100H82O5N2S2, for an intermediate rank coal is shown in Fig. 1 (Zhang et al. 2014, 2015). It was constructed using the Prodrg server (Schuttelkopf and van Aalten 2004). In this model, carbon, hydrogen, oxygen, nitrogen, and sulfur cover about 82.53 %, 5.64 %, 5.5 %, 1.93 %, and 4.4 % of the total mass of the coal, respectively. Constituents and their ratios in this model are similar to that observed in natural coal; therefore, they account for the amorphous and chemically heterogeneous structure of the realistic coal. Generating realistic molecular models of coal is essential for coal simulations applied in coal related research (Mathews et al. 2011).

Building block of intermediate rank coal. Color scheme O red; H white; C cyan; N dark blue; S yellow
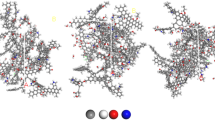
The initial configuration consists of 12 randomly placed coal building blocks (coal molecules) in an empty space of a simulation box which is large enough to accommodate the coal molecules and has x, y, z-dimensions of 3.2 × 3.2 × 3.2 nm3. The simulation box with coal and CO2 is shown in Fig. 2. After the system reaches equilibrium, the volume is around 2.9 × 2.9 × 2.9 nm3 depending on the pressure applied. Obviously, in our system, the void volume (pores within the coal matrix) is at a nanoscale. We connect our system (coal matrix) with an imaginary gas reservoir to allow gas to exchange between the system and the reservoir. The number of gas molecules in the coal matrix varies depending on the pressure or chemical potential applied.

A snapshot for coal and CO2. CO2 molecules are represented by red oxygen and white hydrogen and the rest is the coal
2.2 Molecular model for coal, CO2, CH4, and C2H6
Coal, CO2, CH4, and C2H6 are modeled using the GROMOS force field (Oostenbrink et al. 2004). In this force field, the carbon and the hydrogens that are bonded to it are treated as a single atom, reducing computational effort up to a factor of 9 at the expense of neglecting the slight directional and volume effects of the presence of these hydrogens. Detailed parameter sets can be found in Oostenbrink et al. 2004. In contrast to other biomolecular force fields, this parameterization of the GROMOS force field is based primarily on reproducing the free enthalpies for a range of compounds. The relative free enthalpy is a key property in many biomolecular processes of interest and is why this force field was selected. The non-bonded interactions between atoms which are separated by more than three bonds, or belong to different molecules, are described by pair wise-additive Lennard-Jones (LJ) 12–6 potentials. Cross-interactions between unlike atoms are calculated by the Jorgensen combining rules. The LJ parameters and energy terms and the parameters are taken from Oostenbrink et al. 2004. We use a truncated and shifted potential with a cutoff radius of 14 Å in accordance with the Gromacs force field (Oostenbrink et al. 2004).
2.3 Simulation details
Our simulation procedure consists of MD simulation with a constant number of particles, constant pressure, and constant temperature ensemble (NPT) coupled with MC simulations (Frenkel et al. 1992; Siepmann and Frenkel 1992) in the grand canonical ensemble (GCMC) in which the chemical potentials of the adsorbate, the volume, and the temperature of the system are fixed. The chemical potential (or equivalently the fugacity) is imposed. Instead of setting the chemical potential, it is more intuitive to set the reservoir pressure which is related to the chemical potential by \(\mu = \mu^{0} + RT\ln \left( {\frac{\varphi P}{{p^{0} }}} \right)\), where μ is the chemical potential, and p 0 and μ 0 are the standard pressure and chemical potential, respectively. P is the reservoir pressure and φ is the fugacity coefficient. The temperature T and the chemical potential of the adsorbate phase μ, which is assumed to be in equilibrium with a gas reservoir, are fixed. MD simulation in the NPT ensemble are carried out using Gromacs software (Berendsen et al. 1995; Lindahl et al. 2001; Van der Spoel et al. 2005), while the GCMC algorithm allows the calculation of the isotherm sorption. Periodic boundary conditions are applied in three directions.
In MC, the energy difference between the new configuration and the old configuration is computed (ΔE = E new – E old). If ΔE ≤ 0, the new configuration is accepted. If ΔE > 0, the new configuration is accepted with a Boltzmann-weighted probability of exp(–ΔE/k T), where T is temperature and k is the Boltzmann constant. A more detailed description of the GCMC method can be found in Dubbeldam et al. (2004a, b). To update the configuration, several MC moves are involved. They are translation, swap, and orientation-biased insertions. A translation move is to give a particle a random translation, and the move is accepted or rejected based on the energy difference. A swap move is to insert or delete a particle randomly with a probability of 50 % to allow a chemical equilibrium between the system studied and an imaginary gas reservoir. The orientation-biased insertions are commonly used in MC to insert particles to energetically favorable conformations to increase the acceptance ratio of the moves, especially when density is high for the system under high pressure. Equilibrium is attained when the number of successful insertion and deletion attempts balances each other. The MC simulations are performed using the open source package RASPA 1.0 developed by Dubbeldam et al. (2008). In our simulations, the temperature was fixed using the Berendsen thermostat (Berendsen et al. 1984). As with the temperature coupling, the system can also be coupled to a pressure bath. We use the Berendsen pressure coupling scheme to reach the target pressure, and then switch to Parrinello–Rahman coupling (Parrinello and Rahman 1981) for production runs once the system is in equilibrium as Parrinello–Rahman pressure coupling works in a more efficient and sensible way (Berendsen et al. 1995; Lindahl et al. 2001; Van der Spoel et al. 2005). The equations of motion were integrated with a time step of 0.001 ps. A typical MD production run was ~50 ns. We run MPI parallel programming in the Raijin supercomputer in the National Computational Infrastructure (NCI) Australia and the SGI GPU-based system, Fornax, in iVEC.
3 Simulation results
3.1 Absolute sorption and bulk density
The absolute sorption refers to the actual amount of adsorbate present in the simulation box. Absolute sorption isotherms of CH4, C2H6, and CO2 on dry coal at 308 and 370 K are simulated. Our simulation results for CO2, CH4, and C2H6 plotted in different units of cm3 (STP)/g (Fig. 3a), kg/t (Fig. 3b), and mol/kg (Fig. 3c), show that the absolute sorption of CO2 on dry coal is higher than that of CH4 and C2H6. At 308 K, the initial slope of the absolute sorption of C2H6 is higher than that of CH4 (Fig. 3a and c) due to the stronger affinity between C2H6 and coal and the amount of C2H6 adsorbed in coal is higher than that of CH4 in the low-pressure region. Jiang et al. (1994) showed experimentally that C2H6 sorption isotherm rises sharply and reaches a maximum at a lower pressure than CH4. At high pressure, C2H6 sorption is lower compared with CH4. This observation may be explained by comparing the fugacity of C2H6 and CH4, shown in Fig. 4. The calculated bulk C2H6 fugacity is lower than that of CH4 when pressure is higher than 2 MPa.

Absolute sorption isotherms of CH4, C2H6, and CO2 on coal at a temperature of 308 K in the unit of a cm3 STP/g coal; b kg/t coal; c mol/kg coal

Bulk gas fugacity as a function of bulk gas pressure at 308 K
For high pressure, especially near or above the critical points of the adsorbate, real gas effects must be considered. The Peng–Robinson equation of state (Peng and Robinson 1976) has the general form:
where a and b are constants; P, T, and V are pressure, temperature, and volume, respectively; and R is gas constant. The constants a and b are defined by the following equations:
where T c and P c are critical temperature and pressure, respectively.
Equation (1) defines the compressibility factor (Z):
The compressibility factor measures the deviation from ideal behavior. Z is related to the fugacity (f):
According to Eqs. (2) and (3), we obtain the constant a and b for CH4 and C2H6:
From Eq. (4), positive deviations (Z > 1) are due to the molecules having finite size and is quantified by the constant b; while negative deviations (Z < 1) are due to the molecules having intermolecular forces and are quantified by the constant a. Figure 4 shows negative deviations (below diagonal dash line for ideal gas) for both CH4 and C2H6, indicating that the second term on the right-hand side of Eq. (4) dominates. CH4 and C2H6 deviate from ideal behavior in different ways. Below 2 MPa, they behave similarly. However, above 2 MPa, the deviation of C2H6 from ideal behavior is more significant compared with CH4. This is due to the fact that \(a_{{{\text{C}}_{ 2} {\text{H}}_{ 6} }}\) is 2.4 times of the value of \(a_{{{\text{CH}}_{ 4} }}\). Therefore, we might infer that for hydrocarbon sorption in coal, the constant “a” is identified as an important factor affecting the performance of gas sorption in coal.
In Fig. 5, we show the absolute sorption results for the temperature of 370 K. We observe the same trend for both 308 and 370 K. The absolute sorption (expressed on a mass basis, Figs. 3b and 5b) increases in the order of CH4, C2H6, and CO2. In Figs. 3c and 5c, the absolute molar sorption isotherms of CH4 and C2H6 show characteristic features: they intersect and this takes place at higher pressure when the temperature is higher. The pressure where the sorption curves of CH4 and C2H6 cross shifts from around 1.3 to 3.0 MPa, when temperature is increased from 308 to 370 K. In Fig. 6, we illustrate the temperature effect on the absolute molar sorption for CH4, C2H6, and CO2. It is obvious that the sorption amount decreases with increasing temperature. Other groups also reported that the sorption of gas on coals at a given pressure increases with decreasing temperature (Menon 1968; White et al. 2005; Zhou et al. 2001).

Absolute sorption isotherms of CH4, C2H6, and CO2 on coal at a temperature of 370 K in the unit of a cm3 STP/g coal; b kg/t coal; c mol/kg coal

Temperature effect on the absolute sorption isotherms of a CH4; b C2H6; c CO2
In Fig. 7, we present the bulk density of CH4, C2H6, and CO2 obtained using the Peng–Robinson equation of state (Peng and Robinson 1976). The bulk density increases with pressure but decreases with temperature. The decreases in the bulk density with increasing temperature are simply reflected by the decrease in the sorption capacity when the temperature is raised, shown in Fig. 6. In the case of 308 K, which is close to the critical temperature of C2H6 (305.4 K) and CO2 (304.13 K), there is a sharp increase in the bulk density for both C2H6 and CO2 around their critical pressures. As shown in Fig. 7b, the critical density of C2H6 is 206.18 kg/m3; a clear inflection point appears at the corresponding density at around 5.2 MPa. After the inflection point, one expects the derivative of the bulk density with respect to pressure to decrease. Similarly, in Fig. 7c, we observe an inflection point for CO2 at its critical density of 467.6 kg/m3 at ~8.1 MPa. Bae and Bhatia (2006) have reported an inflection point in the CO2 bulk phase density at a pressure of 8.93 MPa at 313 K. The pressure corresponding to the inflection point increases with temperature. In Fig. 8, we compare the bulk density at 308 K (Fig. 8a) and 370 K (Fig. 8b) for CH4, C2H6, and CO2. The bulk density of CH4 increases linearly with pressure, while the bulk density of C2H6 and CO2 behave non-linearly, especially at 308 K. The linearity of the bulk density with pressure exists up to the pressure of around 4 and 5 MPa for C2H6 and CO2, respectively. Then they go through a significant increase around their inflection point. At the pressure range of 4.9–7.4 MPa, C2H6 has the greatest bulk density followed by CO2 and CH4.

Bulk densities of CH4, C2H6, and CO2 as a function of pressure at 308 and 370 K. a CH4; b C2H6; c CO2

Comparison of the bulk density for CH4, C2H6, and CO2 at a 308 K; b 370 K
Interestingly, we found that the absolute sorption expressed on a volume basis (the absolute sorption in terms of kilogram (kg) per ton (t) of coal, shown in Figs. 3b and 5b, divided by bulk density, presented in Fig. 8a, b) for CH4, C2H6, and CO2 merges with increasing pressure at 308 K (Fig. 9a) and 370 K (Fig. 9b). The Gurvitsch rule (Anderson 1914) predicts that the pore volume occupied by condensable gases and liquids is constant. From our results, it can be inferred that the maximum sorption capacity expressed on volume basis can be described in terms of Gurvitsch’s law. In Fig. 10, we show the temperature effect on the absolute sorption on volume basis for CH4, C2H6, and CO2. As shown in Fig. 10, if pressure is higher than 4 MPa, the temperature dependence of the absolute sorption expressed on a volume basis for CH4, C2H6, and CO2 is negligible, indicating that for each adsorbate, the maximum sorption capacity, expressed on a volume basis, is independent of temperature.

Absolute sorption expressed on a volume basis (adsorbed amount divided by bulk density) for CH4, C2H6, and CO2 as a function of pressure at a 308 K; b 370 K

Temperature effect on the absolute sorption on a volume basis (adsorbed amount divided by bulk density) for a CH4; b C2H6; c CO2
3.2 Excess sorption
Experiments produce excess sorption. The excess sorption is the difference between the absolute sorption and the amount of gas in the reference system. The reference system has the same volume as the sorption system, but the interaction with the solid surface is neglected. The excess molar sorption is given by
where the excess sorption, n e, is the amount in adsorbed phase in excess of the amount that would be present in the pore volume at the equilibrium density of the bulk gas. n a is the absolute amount adsorbed; V p is the pore volume; and ρ b is the equilibrium density of the bulk gas. The pore volume is the volume fraction free to be occupied by gases in sorption processes. In experiments, it is measured using helium, because helium is hardly adsorbed. The point is that helium is a reference gas for measuring excess sorption of all other gases. Whether or not helium actually “adsorbs” is irrelevant. The requirement is that the procedure for measuring pore volume be identical for theoretical prediction and experimental determination. In molecular simulations, void volume is measured by probing the structure with helium at a room temperature of 25 °C. It was obtained from a separate simulation using the Widom particle insertion method. We probed the coal structure with an LJ helium atom at millions of random points, computed the energy difference with and without the particle, and estimated the average Boltzmann weight which directly corresponds to the void fraction (Talu and Myers 2001). In our simulation, all micropores are accessible because of the inherent feature of the MC method. We obtained an average helium pore fraction of 17 % for the dry intermediate rank bituminous coal. Because of the inherent feature of the MC method, the porosity might be overestimated. Based on our porosity result of 17 % and the absolute sorption, we calculate the excess sorption which is the relevant physical observable in experiments.
In Fig. 11, we show the absolute and excess sorption of CH4, C2H6, and CO2 at 308 K. The excess and the absolute quantity are indistinguishable at low pressures in the range up to 0.5 MPa. At higher pressure, the absolute sorption and excess sorption are different. The excess quantity reaches a maximum and then declines. This is due to the fact that excess sorption is relative to what would have been in the pore volume. At high pressures, the bulk phase can still be compressed, but eventually, the pores are filled up and the adsorbed phase density levels off. Once the bulk phase density is higher than the adsorbed phased density, the excess sorption turns negative. The significant increase in the bulk density of C2H6 and CO2 close to their inflection point, shown in Fig. 7, results in a dramatic decrease in the excess sorption of C2H6 and CO2. Inflection points also appear in the excess amount of C2H6 and CO2 at the same pressure as the corresponding inflection point in the bulk density curve, shown in Fig. 7b, c. Li et al. (2010) reported that at 35 °C, the maximum excess sorption capacity of the medium-volatile bituminous coal (dry, ash-free) for CH4 and CO2 is around 0.8 and 1.3 mol/kg, respectively. Our molecular simulation results indicate a maximum excess sorption capacity of around 0.82 and 1.57 mol/kg for CH4 and CO2 on the bituminous coal (dry, ash-free), respectively. The results of the maximum excess sorption for CH4 and CO2 reported by Li et al. (2010) are around 2.4 % and 17.2 % lower than our simulation results. This could be attributed to the fact that the porosity might be overestimated due to the inherent feature of the MC method. A good agreement between the experiment and the molecular simulation indicates that the sorption mainly takes place in the coal matrix.

Absolute and excess sorption of CH4, C2H6, and CO2 on coal at 308 K. a CH4; b C2H6; c CO2
In Fig. 12, we show the temperature effect on the excess sorption of CH4, C2H6, and CO2. Similar to those obtained by Li et al. (2010), we also observed that after passing through the maximum, the lower temperature excess sorption isotherms decline more rapidly than the higher temperature isotherms, which results in an intersection of the isotherms at ~4.5 MPa for C2H6 and ~7.8 MPa for CO2. The intersection corresponds to a reversal point of the temperature dependence of the excess sorption isotherms. At the pressure range above the reversal point, in contrast to the low-pressure range, the excess sorption increases with temperature. This effect is clearly related to the bulk density change of the C2H6 and CO2, shown in Fig. 8. It is evident that for high pressures, the bulk density is much higher at lower temperature, and the excess amount reduces more quickly. When the bulk phase density approaches the adsorbed phase density, the excess sorption would become zero.

Temperature effect on the excess sorption of CH4, C2H6, and CO2 on coal. a CH4; b C2H6; c CO2
Figure 13 compares the ratio of absolute and excess adsorbed amounts of CO2–CH4 on coal at 308 K. The ratio for both the absolute and excess amount decreases with increasing pressure. Above 8 MPa, the ratio of the excess amounts adsorbed at 308 K is less than one, suggesting that pressures lower than 8 MPa may provide efficient CO2 sequestration and methane replacement in coal bed. Bae and Bhatia (Bae and Bhatia 2006) suggested an optimum pressure of 10 MPa at a higher temperature of 313 K. Similarly, we present the ratio of the absolute and excess adsorbed amounts of CO2–C2H6 on coal at 308 K in Fig. 14. The ratio of the absolute adsorbed amounts increases when pressure is less than l MPa and then levels off around an average value of 2, but the ratio of the excess adsorbed amounts increases sharply when pressure is over 4 MPa, where the excess adsorbed amount of C2H6 drops significantly, suggesting an optimum pressure of above 4 MPa for C2H6 replacement.

Ratio of the absolute and excess adsorbed amounts of CO2–CH4 on coal at 308 K

Ratio of the absolute and excess adsorbed amounts of CO2–C2H6 on coal at 308 K
3.3 Interaction energy and radial distribution functions (RDFs)
We further investigate the sorption mechanism by analyzing the interaction energy between the coal and adsorbate. We compare the interaction energy of adsorbate–adsorbate and adsorbate–coal. The results are given in Fig. 15. For the purpose of clarity, only the results at 308 K are shown. The interaction energy between the coal and CO2 is much higher than between the coal and hydrocarbons. The interaction energy between the coal and the CO2 becomes systematically more negative with increasing pressure. The increasingly negative energies signify greater interactions between the coal and CO2 molecules with an increasing pressure. Based on the observation that the coal-CO2 interaction is ~2.2–4.5 times of the interaction energy of coal-CH4 and ~1.3–2.2 times of coal-C2H6. We infer that the higher sorption of CO2 is mainly caused by stronger intermolecular interactions between coal and CO2.

Interaction energy of coal–CO2, coal–C2H6, coal–CH4, CO2–CO2, C2H6–C2H6, and CH4–CH4 at 308 K
RDFs are defined as the ratio of the number of atoms at a distance r from a given atom compared with the number of atoms at the same distance in an ideal gas with the same density. The peak and the shape of the RDFs can reflect the density and structure of the system. To further investigate the effect of packing of the adsorbate on the sorption, we compare the RDFs of CH4–CH4, C2H6–C2H6, and CO2–CO2 at 308 K and 10 MPa in Fig. 16. As it is shown, the distance of closest contact in the O–O, O–C, and C–C RDFs of CO2 is ~2.5 Å, but for hydrocarbons, the closest contact appears at 3.2 Å. It indicates that the distance between CO2 molecules is shorter and CO2 molecules are more closely packed compared with CH4 and C2H6. We found the first contact peak between CH4 and CH4 is more significant than between C2H6 and C2H6, but at a separation distance of ~4.4–6.9 Å, the curve of the C2H6–C2H6 RDF is above that of CH4–CH4, indicating that the separation between C2H6 molecules is larger than the distance between the CH4 molecules and C2H6 is loosely packed compared with CH4.

Radial distribution functions (RDFs) of CH4, C2H6, and CO2 at 308 K and 10 MPa
4 Conclusion
We have performed the combined MD and MC simulations on CH4, C2H6, and CO2 sorption on dry intermediate rank coal in a pressure range up to 10 MPa and at temperatures of 308 and 370 K (35 and 97 °C). Our results indicate that absolute sorption (expressed as a mass basis) divided by bulk density is independent of temperature for both hydrocarbons (CH4 and C2H6) and CO2 when pressure is over 4 MPa. We infer that temperature has negligible effect on the maximum absolute sorption on a volume basis. We also observe that the intermediate rank coal has close maximum sorption capacity expressed on a volume basis for CH4, C2H6, and CO2. Based on the observation, we could infer that the pore volume occupied by CH4, C2H6, and CO2 is similar and the sorption capacity expressed on volume basis can be described in terms of Gurvitsch’s law. The comparisons of our adsorption isotherm obtained from molecular simulation with published experimental data are satisfactory. It indicates that the CH4, C2H6, and CO2 are mainly stored in a coal matrix by sorption. Our results reveal that the “a” constant (proportional to T 2 c /P c ) in the Peng–Robinson equation of state is an important factor affecting the sorption behavior of hydrocarbons. This study provides a quantitative understanding of the effect of temperature on CH4, C2H6, and CO2 sorption capacity from a microscopic perspective. It also offers insights into aspects of the excess sorption and its relationship with bulk phase density. Molecular simulation proves to be a cost-effective and efficient method for directly studying the interactions between coal and gases under various external environments and for predicting gas sorption behavior in complicated and complex systems.
References
Anderson JS. Structure of silicic acid gels. Z Physik Chem. 1914;88:191–228.
Azmi AS, Yusup S, Muhamad S. The influence of temperature on adsorption capacity of Malaysian coal. Chem Eng Process. 2006;45(5):392–6.
Bae JS, Bhatia SK. High-pressure adsorption of methane and carbon dioxide on coal. Energy Fuels. 2006;20(6):2599–607.
Berendsen HJC, Postma JPM, Vangunsteren WF, et al. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–90.
Berendsen HJC, Vanderspoel D, Vandrunen R. Gromacs: a message-passing parallel molecular-dynamics implementation. Comput Phys Commun. 1995;91(1–3):43–56.
Billemont P, Coasne B, De Weireld G. An experimental and molecular simulation study of the adsorption of carbon dioxide and methane in nanoporous carbons in the presence of water. Langmuir. 2011;27(3):1015–24.
Billemont P, Coasne B, De Weireld G. Adsorption of carbon dioxide, methane, and their mixtures in porous carbons: effect of surface chemistry, water content, and pore disorder. Langmuir. 2013;29(10):3328–38.
Brochard L, Vandamme M, Pelenq RJM, et al. Adsorption-induced deformation of microporous materials: coal swelling induced by CO2–CH4 competitive adsorption. Langmuir. 2012a;28(5):2659–70.
Brochard L, Vandamme M, Pellenq RJM. Poromechanics of microporous media. J Mech Phys Solids. 2012b;60(4):606–22.
Busch A, Gensterblum Y. CBM and CO2-ECBM related sorption processes in coal: a review. Int J Coal Geol. 2011;87(2):49–71.
Busch A, Gensterblum Y, Krooss BM. Methane and CO2 sorption and desorption measurements on dry Argonne premium coals: pure components and mixtures. Int J Coal Geol. 2003;55(2–4):205–24.
Busch A, Gensterblum Y, Krooss BM, et al. Methane and carbon dioxide adsorption-diffusion experiments on coal: upscaling and modeling. Int J Coal Geol. 2004;60(2–4):151–68.
Connell LD, Lu M, Pan ZJ. An analytical coal permeability model for tri-axial strain and stress conditions. Int J Coal Geol. 2010;84(2):103–14.
Day S, Fry R, Sakurovs R. Swelling of Australian coals in supercritical CO2. Int J Coal Geol. 2008;74(1):41–52.
Dubbeldam D, Calero S, Ellis D, et al. RASPA 1.0: Molecular Software Package for adsorption and diffusion in nanoporous materials. Evanston: Northwestern University; 2008.
Dubbeldam D, Calero S, Vlugt TJH, et al. Force field parametrization through fitting on inflection points in isotherms. Phys Rev Lett. 2004a;93(8):088302.
Dubbeldam D, Calero S, Vlugt TJH, et al. United atom force field for alkanes in nanoporous materials. J Phys Chem B. 2004b;108(33):12301–13.
Fitzgerald JE, Pan Z, Sudibandriyo M, et al. Adsorption of methane, nitrogen, carbon dioxide and their mixtures on wet Tiffany coal. Fuel. 2005;84(18):2351–63.
Frenkel D, Mooij GCAM, Smit B. Novel scheme to study structural and thermal-properties of continuously deformable molecules. J Phys Condens Matter. 1992;4(12):3053–76.
Gensterblum Y, Busch A, Krooss BM. Molecular concept and experimental evidence of competitive adsorption of H2O, CO2 and CH4 on organic material. Fuel. 2014;115:581–8.
Goodman AL, Busch A, Bustin RM, et al. Inter-laboratory comparison II: CO2 Isotherms measured on moisture-equilibrated Argonne premium coals at 55 °C and up to 15 MPa. Int J Coal Geol. 2007;72(3–4):153–64.
Jiang SY, Zollweg JA, Gubbins KE. High-pressure adsorption of methane and ethane in activated carbon and carbon-fibers. J Phys Chem-Us. 1994;98(22):5709–13.
Krooss BM, van Bergen F, Gensterblum Y, et al. High-pressure methane and carbon dioxide adsorption on dry and moisture-equilibrated Pennsylvanian coals. Int J Coal Geol. 2002;51(2):69–92.
Li DY, Liu Q, Weniger P, et al. High-pressure sorption isotherms and sorption kinetics of CH4 and CO2 on coals. Fuel. 2010;89(3):569–80.
Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7(8):306–17.
Lu M, Connell LD. A model for the flow of gas mixtures in adsorption dominated dual porosity reservoirs incorporating multi-component matrix diffusion - Part I.Theoretical development. J Petrol Sci Eng. 2007;59(1–2):17–26.
Lu M, Connell LD, Pan ZJ. A model for the flow of gas mixtures in adsorption dominated dual-porosity reservoirs incorporating multi-component matrix diffusion-Part II numerical algorithm and application examples. J Petrol Sci Eng. 2008;62(3–4):93–101.
Mathews JP, van Duin ACT, Chaffee AL. The utility of coal molecular models. Fuel Process Technol. 2011;92(4):718–28.
Menon PG. Adsorption at high pressures. Chem Rev. 1968;68(3):277–94.
Oostenbrink C, Villa A, Mark AE, et al. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25(13):1656–76.
Ottiger S, Pini R, Storti G, et al. Competitive adsorption equilibria of CO2 and CH4 on a dry coal. Adsorption. 2008;14(4–5):539–56.
Pan ZJ, Connell LD. A theoretical model for gas adsorption-induced coal swelling. Int J Coal Geol. 2007;69(4):243–52.
Pan ZJ, Connell LD. Comparison of adsorption models in reservoir simulation of enhanced coalbed methane recovery and CO2 sequestration in coal. Int J Greenhouse Gas Control. 2009;3(1):77–89.
Pan ZJ, Connell LD. Modelling permeability for coal reservoirs: a review of analytical models and testing data. Int J Coal Geol. 2012;92:1–44.
Parrinello M, Rahman A. Polymorphic transitions in single-crystals: a new molecular-dynamics method. J Appl Phys. 1981;52(12):7182–90.
Peng D, Robinson DB. New 2-constant equation of state. Ind Eng Chem Fund. 1976;15(1):59–64.
Pini R, Ottiger S, Storti G, et al. Pure and competitive adsorption of CO2, CH4 and N2 on coal for ECBM. Greenhouse Gas Control Technologies 9. 2009; 1(1): 1705–10.
Pini R, Ottiger S, Storti G, et al. Prediction of competitive adsorption on coal by a lattice DFT model. Adsorption. 2010;16(1–2):37–46.
Sakurovs R, Day S, Weir S. Relationships between the critical properties of gases and their high pressure sorption behavior on coals. Energy Fuels. 2010;24:1781–7.
Sakurovs R, Day S, Weir S, et al. Application of a modified Dubinin–Radushkevich equation to adsorption of gases by coals under supercritical conditions. Energy Fuels. 2007;21(2):992–7.
Schuttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D-Biol Crystallogr. 2004;60:1355–63.
Siepmann JI, Frenkel D. Configurational Bias Monte-Carlo: a new sampling scheme for flexible chains. Mol Phys. 1992;75(1):59–70.
Spiro CL, Kosky PG. Space-Filling Models for Coal. 2. Extension to Coals of Various Ranks. Fuel. 1982;61(11):1080.
Talu O, Myers AL. Molecular simulation of adsorption: gibbs dividing surface and comparison with experiment. AIChE J. 2001;47(5):1160–8.
Tambach TJ, Mathews JP, van Bergen F. Molecular exchange of CH4 and CO2 in coal: enhanced coalbed methane on a nanoscale. Energy Fuels. 2009;23:4845–7.
Van der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–18.
Vandamme M, Brochard L, Lecampion B, et al. Adsorption and strain: the CO2-induced swelling of coal. J Mech Phys Solids. 2010;58(10):1489–505.
White CM, Smith DH, Jones KL, et al. Sequestration of carbon dioxide in coal with enhanced coalbed methane recovery: a review. Energy Fuels. 2005;19(3):659–724.
Zhang J, Clennell MB, Dewhurst DN, et al. Combined Monte Carlo and molecular dynamics simulation of methane adsorption on dry and moist coal. Fuel. 2014;122:186–97.
Zhang J, Liu K, Clennell MB, et al. Molecular simulation of CO2–CH4 competitive adsorption and induced coal swelling. Fuel. 2015;160:309–17.
Zhou L, Zhou YP, Bai SP, et al. Determination of the adsorbed phase volume and its application in isotherm modeling for the adsorption of supercritical nitrogen on activated carbon. J Colloid Interface Sci. 2001;239(1):33–8.
Acknowledgments
We thank the National Computing Infrastructure (NCI) national facility and iVEC GPU cluster for a generous allocation of computing time. The second author was supported by the National Basic Research Program of China (2014CB239004) and the “Element and Process Constraint Petroleum System Modeling” project (No. 2011A-0207) under the PetroChina Science Innovation program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Xiu-Qin Zhu
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, J., Liu, K., Clennell, M.B. et al. Molecular simulation studies of hydrocarbon and carbon dioxide adsorption on coal. Pet. Sci. 12, 692–704 (2015). https://doi.org/10.1007/s12182-015-0052-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12182-015-0052-7