
Abstract
The present study primarily focuses on the efficacy of Malabaricone C (Mal C) as an anti-inflammatory agent. Mal C inhibited mitogen-induced T-cell proliferation and cytokine secretion. Mal C significantly reduced cellular thiols in lymphocytes. N-acetyl cysteine (NAC) restored cellular thiol levels and abrogated Mal C-mediated inhibition of T-cell proliferation and cytokine secretion. Physical interaction between Mal C and NAC was evinced from HPLC and spectral analysis. Mal C treatment significantly inhibited concanavalin A-induced phosphorylation of ERK/JNK and DNA binding of NF-κB. Administration of Mal C to mice suppressed T-cell proliferation and effector functions ex vivo. Mal C treatment did not alter the homeostatic proliferation of T-cells in vivo but completely abrogated acute graft-versus-host disease (GvHD)-associated morbidity and mortality. Our studies indicate probable use of Mal C for prophylaxis and treatment of immunological disorders caused due to hyper-activation of T-cells.
Similar content being viewed by others

1 Introduction
Malabaricone C (Mal C), derived from plants of the family Myristicaceae (dried fruit arils of Myristica fragrans and Myristica malabarica), has been shown to possess several biological properties in different experimental systems (Patro et al. 2005, 2010; Banerjee et al. 2008; Chong et al. 2011; Tyagi et al. 2014a,b). Mal C, a potential pharmacological ingredient, is an antioxidant and is present as a bioactive component in several traditional medicinal preparations from India (in Ayurveda) and China (Patro et al. 2005).
The chemical similarity of Mal C to other malabaricones (Mal A, Mal B, and Mal D) present in plant material makes its isolation tedious. To cater to the need of copious amounts of Mal C for an in-depth study of its pharmacological profile, a novel method for efficient chemical synthesis of Mal C by adopting a cross-metathesis strategy was optimized in our laboratory (Kundu and Nayak 2017). Mal C is reported to inhibit lipopolysaccharide (LPS)-induced inflammation in macrophages by altering cellular redox and inhibition of NF-κB (Kang et al. 2012). Our previous findings demonstrated that cellular redox modulation can be used as an effective strategy for the suppression of T-cell hyper-activation (Checker et al. 2009; Patwardhan et al. 2011, 2016; Gambhir et al. 2014). The present studies were undertaken to explore whether Mal C could modulate T-cell responses in vitro and in vivo via redox modulation.
Activation of antigen-specific T-cells by antigen-presenting cells bearing cognate antigen-loaded MHC molecules is essential for the orchestration of adaptive immune response. The activated T-helper cells proliferate and differentiate into either Th1, Th2, Th17, Tfh, or Treg cells and mediate responses against intracellular pathogens, and participate in intracellular defence, clearance of highly pathogenic bacteria/fungus, activation of antibody production in B-cell follicles, and suppression of autoimmunity (Gagliani and Huber 2017).
The proliferation and differentiation of T-cells following interaction with cognate peptide-MHC-bearing antigen-presenting cells are influenced by the signal strength, signal duration, co-stimulation, and cytokines produced by the antigen-presenting cells (APCs) (Gett et al. 2003; van Stipdonk et al. 2003). The extracellular redox environment as well as the reactive oxygen species present inside T-cells also play a causal role in shaping T-cell-mediated immunity (Gelderman et al. 2006; Patwardhan et al. 2016). T-cells require a reducing environment and specific production of mitochondrial reactive oxygen species for their activation and proliferation (Sena et al. 2013; Simeoni and Bogeski 2015). Alterations in the redox balance can affect their differentiation and immune response. Dysregulated T-cell activation is associated with many inflammatory disorders including COVID-19, autoimmunity, psoriasis, rheumatoid arthritis, graft rejection, GvHD, etc. (Griffiths 2005; Kesarwani et al. 2013; Suh et al. 2014). Herein, we performed experiments to evaluate the effect of Mal C on T-cell activation, proliferation, cytokine production, and differentiation. In the present study, we attempted to identify a causal role of redox modulation in suppression of T-cells responses by Mal C and evaluate its efficacy as a prophylactic agent for acute GvHD without perturbing T-cell homeostasis.
2 Materials and methods
2.1 Chemical compound
Malabaricone C [IUPAC name: 1-(2,6-dihydroxyphenyl)-9-(3,4-dihydroxyphenyl)nonan-1-one)] used in this study was synthesized according to our previously reported method (Kundu and Nayak 2017). The Mal C so synthesized was fully characterized by 1H NMR (supplementary figure 3) and 13C NMR spectroscopy (supplementary figure 4) (Kundu and Nayak 2017). The purity of Mal C was estimated by HPLC analysis. The major peak area of Mal C was found to be >95% of the combined total peak area (supplementary figure 5)
2.2 Reagents
Concanavalin A (Con A) was procured from Calbiochem (Darmstadt, Germany). Propidium iodide (PI), triethanolamine, vinylpyridine, 5-sulfosalicylic acid, NADPH, tritonX100, 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB), boric acid, polyacrylamide, TEMED, 2-mercaptoethanol, ammonium per sulfate, SDS, sodium azide, sodium deoxycholate, NP40, aprotinin, benzamidine, leupeptin, PMSF, methanol, monochlorobimane (MCB), EDTA, Tris-HCl, dimethylsulfoxide, NaCl, protease inhibitor cocktail, immunoblot PVDF membrane, dihydrodichlorofluoroscein diacetate (H2DCFDA), N-acetyl cysteine (NAC), glutathione, low melting agarose, enhanced chemiluminescence substrate (POD), HPLC grade acetonitrile (ACN), and formic acid were obtained from Sigma-Aldrich (MO, USA). Bradford reagent, molecular weight marker, fetal bovine serum (FBS), and RPMI 1640 were purchased from HiMedia (Mumbai, India). Carboxyfluorescein diacetate succinimidyl ester (CFSE) was purchased from Molecular Probes (NY, USA). CD4+ T-cell isolation kit was procured from Miltenyi Biotech. IL-2, IL-6, and IFN-γ ELISA sets, monoclonal antibodies (mAb) against CD3/CD28, fluorochrome conjugated antibodies against CD69, CD25, pERK, and pJNK were all purchased from BD Pharmingen (CA, USA). pERK, pJNK, ERK, and JNK antibodies for western blotting were purchased from Cell Signaling Technologies (Boston, MA). Oligonucleotide probe of NF-κB was purchased from Santacruz Biotechnology (Dallas, Texas, USA). T4 kinase buffer and T4 polynucleotide kinase were obtained from New England Biolabs (Ipswich, MA). All other reagents and chemicals used in this study were of analytical grade.
2.3 Animal studies
Swiss, BALB/c, and C57BL/6 male mice (8–10 weeks old; weighing 20–25 g) used in this study were obtained from the animal house facility of Radiation Biology and Health Sciences Division of Bhabha Atomic Research Centre (BARC). Institutional Animal Ethics Committee guidelines were strictly followed.
2.4 Proliferation assay and CD4+ T-cell sorting
Lymphocytes were isolated and stained with CFSE (5 μM) as described previously (Checker et al. 2009). The lymphocytes were incubated with Mal C (1 to 10 μM, 2 h) with or without the pre-treatment of NAC or GSH for 2 h, and stimulated with Con A (2.5 μg/mL) or surface bound anti-CD3 mAb (1 μg/mL)/soluble anti-CD28 mAb (0.5 μg/mL) for 72 h at 37°C with 10% FBS in a 95% air/5% CO2 atmosphere. Cells treated with vehicle served as control. Cell proliferation was measured by dye dilution using a flow cytometer (Partec CyFlow). Cells displaying reduction in fluorescence intensity of CFSE were estimated using FlowJo software (Treestar Inc, Ashland, USA) and plotted as the percentage of proliferating cells.
CD4+ T-cell isolation from splenic lymphocytes was carried out following the manufacturer’s protocol using magnetic sorting. Briefly, 10×106 cells were re-suspended in buffer containing a cocktail of biotin-conjugated monoclonal antibodies against CD8a, CD11b, CD11c, CD19, CD45R (B220), CD49b (DX5), CD105, Anti-MHC Class II, Ter-119, and TCRγ/δ, followed by the addition of magnetic nanoparticle-conjugated streptavidin-PE. Cells were placed under a magnetic field, and magnetically labelled cells remained in the magnet while unbound CD4+ T-cells were collected. Purity was assessed by staining cells with anti-CD4 antibody.
2.5 Estimation of cytokines
Lymphocytes (1×106) treated as described above were cultured for 24 h and the concentration of cytokines (IL-2, IL-6, and IFN-γ) was measured in culture supernatant using ELISA.
2.6 Surface staining and intracellular staining
Cells were stained with fluorochrome (PE or FITC)-conjugated mAbs as previously described (Sharma et al. 2012). Briefly, lymphocytes (2.5×106) were incubated with Mal C (10 μM, 2 h) followed by stimulation with Con A (2.5 μg/mL, 24 h for surface staining, and 2.5 μg/mL, 2 h for intracellular staining) and subsequent staining with fluorochrome-conjugated CD69 or CD25 monoclonal antibodies. Unstained and isotype-stained cells were used as controls. Then, cells were acquired on a BD FACS Melody flow cytometer and analyzed using FlowJo software.
2.7 Propidium iodide staining
Cells were washed with 1X PBS and the pellet was resuspended in propidium iodide (PI) staining solution (1X PBS containing sodium citrate, RNase A and 0.1% triton X 100) overnight at 4°C. Cells were acquired using Highthroughput Cell Analyzer (SPT Labtech, UK) and analyzed for hypo-diploid cell percentage.
2.8 Estimation of ROS and GSH
For ROS estimation based on DCF fluorescence (excitation/emission at 485/535 nm), Mal C- or vehicle-treated cells were stained with H2DCF-DA and monitored using a spectrofluorimeter (Synergy H1 Hybrid Microplate Reader, BioTek). For quantitation of GSH (reduced/oxidized) levels, cells were treated with Mal C or vehicle control, harvested, and lysed, and the cell extract was probed for GSH quantitation as previously described (Patwardhan et al. 2015). For estimation of cellular thiols, cells were stained with MCB (excitation/emission at 390/478 nm), followed by flow cytometric acquisition.
2.9 HPLC analysis of Mal C and NAC interaction
We employed a multi-step gradient mobile phase with acetonitrile as solvent A and aqueous formic acid (0.1%) as solvent B to carry out chromatographic separation using a C-18 column with an optimized method. The gradient was set as: 40% solvent A for 2 min; 80% solvent A for 22 min; 100% solvent A for 2 min; 40% solvent A for 4 min, and the flow rate was maintained at 1 mL/min. The sample volume for injection was 20 μL and the total run time was 30 min and Mal C was detected at a retention time (Rt) of 15.773 min. Detection was carried out using a diode array detector at 274 nm. A stock solution of NAC of 100 mM concentration was prepared and dilution was done in 1X PBS. Mal C was added to NAC (Mal C 100 μM + NAC 100 µM) and was incubated at 37°C for 1 h and run on HPLC (Dionex Ultima 3000 series HPLC system with Chromeleon software (version 6.8)).
2.10 Spectroscopic analysis of Mal C and NAC interaction
Mal C (100 μM) was incubated with NAC (100 μm) in water at 37°C for 1 h, and absorbance spectra were recorded from 250 to 700 nm using a Jasco Spectrophotometer. A graph showing absorbance versus wavelength (nm) was plotted using Jasco Spectra Manager Ver. 2 software.
2.11 Western blotting
Cells (10×106/mL) treated with Mal C or Con A (2.5 μg/mL) or both were harvested, and lysed using 1X RIPA lysis buffer consisting of 50 mM Tris HCl, 150 mM NaCl, 1.0% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 1.0 mM EDTA, 0.1% (w/v) SDS and 0.01% (w/v) sodium azide, protease inhibitor cocktail, and phosphatase inhibitor at a pH of 7.4. Protein estimation was carried out using the Bradford assay followed by electrophoretic separation on SDS-PAGE (10%). Proteins were transferred onto a PVDF membrane and probed for pERK, pJNK, ERK, and JNK using monoclonal antibodies followed by the HRP-conjugated secondary antibody. The bands were visualized in SynGene: GBox (Cambridge, UK) Gel documentation system using an enhanced chemiluminescence kit (POD).
2.12 Gel shift assay for transcription factor
Cells (10×106/mL) were treated with vehicle or indicated concentrations of Mal C or Con A (2.5 μg/mL) or both. Nuclear extracts were prepared from the cells and incubated with 32P-end-labeled 45-mer double-stranded NF-κB oligonucleotides from the human immunodeficiency virus long terminal repeat (5′-TTGTTACAAGGGACTTTCCGCTGGGGACTTTCCAGGGAGGCGTGG-3′) as previously described (Patwardhan et al. 2015, 2016). The sample was run on a native polyacrylamide gel followed by drying of the gel and its subsequent exposure to a phosphor image plate for visualizing radioactive bands using a PhosphorImage plate scanner (Amersham Biosciences, USA).
2.13 Ex vivo stimulation of lymphocytes
The high lipophilicity and low water solubility of Mal C leads to poor oral bioavailability, and hence mice were administered with Mal C (10 mg/kg body weight) or vehicle intra-peritoneally (3 mice per group) and were sacrificed 24 h after the injection. Lymphocytes were stimulated with Con A (2.5 μg/mL) for 24 h for estimation of secreted cytokines using ELISA. Another set of lymphocytes was stained with CFSE and stimulated with Con A (2.5 μg/mL) for 72 h for assessment of cell proliferation by flow cytometry.
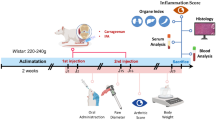
2.14 Graft-versus-host disease
Recipient BALB/c mice were exposed to whole-body irradiation (WBI) of 6 Gy at a dose rate of 1 Gy/min in a blood irradiator (BRIT, Vashi, India) for induction of lymphopenia (6 mice per group). The lymphocytes from allogenic C57BL/6 donors were treated with vehicle control or Mal C in vitro, and 10 million cells were administered intravenously into the lateral tail vein of lymphopenic recipients 48 h after WBI. After engraftment, lymphopenic recipient mice were monitored for assessment of changes in the body weight and survival up to 30 days. On day 5 post engraftment, blood was collected from lymphopenic recipient mice and the serum was separated to monitor the cytokine levels using ELISA.
2.15 Monitoring homeostatic proliferation in vivo
CD4+ T-cells were sorted from lymphocytes of BALB/c mice using magnetic beads and were stained with CFSE. The cells were incubated with Mal C (10 μM) or vehicle for 4 h at 37°C in 5% CO2. The cells were washed and 1 million CFSE+ CD4+ T-cells were intravenously engrafted into lymphopenic syngeneic BALB/c mice (3 mice per group). After 72 h, lymphocytes from lymphopenic recipient mice were isolated and the frequency and proliferation of donor CFSE+ cells were monitored by flow cytometry.
2.16 Statistics
To calculate statistical significance between groups, Student’s t-tests were performed. For more than two groups, one-way ANOVA followed by Tukey’s multiple comparison tests were performed using GraphPad Prism 6.0 software. The p-values are displayed in the respective figures. Data points represented in the study indicate means±SEM. Survival curve comparisons were conducted using the log-rank (Mantel-Cox) test.
3 Results
3.1 Mal C inhibited T-cell activation, proliferation, and cytokine production
The cellular redox state of a T-cell can affect T-cell receptor signaling. ROS are important for balancing T-cell activation versus inactivation, thereby regulating immune outcomes. Mal C is a redox-active phytochemical, and hence we assessed the effect of Mal C treatment on T-cell responses. Mal C treatment suppressed the proliferation of Con A-stimulated T-cells as evinced from the reduction in the percentage of proliferating cells (figure 1A and B) and effector cytokines (IL-2 and IFN-γ) secretion in activated T-cells (figure 1C). Mal C inhibited the proliferation of αCD3/αCD28-activated T-cells (figure 1D and E) and suppressed the secretion of IL-2 and IFN-γ (figure 1F). Further, Mal C treatment also suppressed αCD3/αCD28 mAb-induced proliferation of CD4+ T-cells (figure 1G) and IL-2 secretion (supplementary figure 1D). However, Mal C did not compromise the viability of unstimulated and stimulated lymphocytes (supplementary figure 1A–C).

Mal C significantly inhibited the proliferation and cytokine secretion induced by Con A as well as anti-CD3/anti-CD28 mAb in vitro: Lymphocytes stained with CFSE, and incubated with varying concentrations of Mal C (1, 5 and 10 μM) for 2h, were subsequently stimulated with either Con A (2.5 μg/mL) or anti-CD3/CD28 mAb and then cultured for 72 h or 24 h at 37°C, respectively. These lymphocytes were then acquired using a flow cytometer and the overlaid flow cytometric histograms are shown in (A) and concatenated flow cytometric histograms are shown in (D). Percentage of proliferating cells was estimated using FlowJo based on CFSE dye dilution and graphed in (B) and (E). Estimation of cytokine levels (IL-2 and IFN-γ) was carried out from the culture supernatants obtained from lymphocytes cultured for 24 h. Cytokine concentration in pg/mL is graphed in (C) and (F). Magnetically isolated, CFSE stained, CD4+ T-cells were treated with vehicle or Mal C, stimulated with anti-CD3/anti-CD28 mAb and acquired using flow cytometer. The bar graph represents the percentage of proliferating cells (G). Three independent experiments were performed with data points representing means±SEM from three replicates from one such experiment.
3.2 Mal C suppressed mitogen-induced activation of cell surface markers, MAPK and NF-κB
The expression of T-cell activation markers CD69 and CD25 in Con A-activated lymphocytes was estimated at 24 h by antibody staining and flow cytometry. Mal C significantly suppressed CD69 (figure 2A and B) as well as CD25 (figure 2C and D) expression in mitogen-activated T-cells, indicating its interference with T-cell signaling. We investigated the effect of Mal C on mitogen-induced phosphorylation of MAP kinases such as ERK and JNK. Mal C treatment inhibited activation of ERK (figure 2E and F) and JNK (figure 2G and H) in lymphocytes following mitogenic stimulation. Further, nuclear levels of NF-κB were estimated and it was observed that Mal C significantly inhibited the DNA binding of the NF-κB induced after mitogenic stimulus (figure 2I and J).

Mal C suppressed the expression of T-cell activation markers, mitogen-induced phosphorylation of MAPK, and NF-κB DNA binding: Lymphocytes (2.5×106) incubated with Mal C (10 μM) followed by stimulation with Con A (2.5 μg/mL) were subsequently cultured for 24 h at 37°C and then stained with anti-CD69-FITC (A and B) or anti-CD25-PE (C and D) conjugated antibodies. Lymphocytes stained in such manner were then acquired using a flow cytometer, and concatenated flow cytometric histograms are shown. The percentage of positive cells are graphed in (B) and (D). Three independent experiments were performed with data points representing mean±SEM from three replicates from one such experiment. Lymphocytes incubated with vehicle or Mal C (1 h) were treated with Con A (2.5 μg/mL for 3 h) followed by probing for pERK and ERK (E and F), and pJNK and JNK (G and H) in the whole-cell lysates by western blotting. The bar graphs show quantification of blots from three independent experiments (F and H). Nuclear extracts were prepared from lymphocytes incubated with Mal C 10 μM for 1 h followed by stimulation with Con A (2.5 μg/mL) for 4 h and subsequently probed for NF-κB DNA binding by EMSA (I). The bar graph in (J) shows the quantitation of NF-κB band intensity from three independent experiments.
3.3 Mal C modulated cellular redox status in lymphocytes
To evaluate the effect of Mal C on cellular redox status in murine lymphocytes, the levels of reactive oxygen species (ROS) were measured by DCFDA staining. Mal C treatment significantly suppressed ROS levels in resting as well as activated cells (figure 3A) and decreased cellular GSH/GSSG ratio (figure 3B).

Mal C modulated cellular redox, and thiol antioxidants abrogated its anti-inflammatory effects: Cells were treated with vehicle or Mal C (2 h) followed by stimulation with Con A (30 min). Unstimulated or stimulated cells were stained with H2DCFDA, and subsequently change in fluorescence was monitored using a flow cytometer. The bar graph shows mean fluorescence intensity (MFI) (A). Lymphocytes incubated with Mal C (10 μM) for the indicated time points were processed for estimation of cellular GSH. The GSH/GSSG ratio is graphed in (B). Lymphocytes stained with CFSE were incubated with thiol antioxidants NAC (10 mM) or GSH (10 mM) for 2 h prior to incubation with Mal C. Following mitogenic stimulation by Con A for 72 h, cells were acquired using a flow cytometer, and the concatenated flow cytometric histogram is shown in (C). The percentage of proliferating cells is graphed in (D). Culture supernatants harvested at 24 h were subjected to estimation of cytokines using ELISA. The cytokine concentration (pg/mL) is graphed in (E). Three independent experiments were performed with data points representing mean±SEM from three replicates from one such experiment. Mal C interacted with N-acetyl cysteine (NAC): Mal C (10 μM) was incubated with NAC (100 μM) at 37°C for 1 h in 1X PBS and run on HPLC. The HPLC chromatogram is shown in (F). NAC reversed Mal C-induced depletion of cellular thiols: Cells incubated with Mal C or NAC or both for indicated time points were stained with monochlorobimane (MCB) and subsequently acquired using a flow cytometer for monitoring change in fluorescence, and the concatenated flow cytometric histograms are shown in (G). MCB mean fluorescence intensity is graphed in (H). Three independent experiments were performed with data points representing means±SEM from three replicates in each experiment.
To identify the role of cellular redox in the immunosuppressive action of Mal C, lymphocytes stained with CFSE were incubated with thiol (SH)-containing antioxidants (GSH/NAC 10 mM) followed by stimulation with mitogen in the presence of Mal C. Mal C-mediated suppression of T-cell proliferation (figure 3C and D) and IL-2 and IFN-γ secretion (figure 3E) were abrogated by thiol-containing antioxidants.
Mal C was incubated with different concentrations of NAC for 1 h at 37°C and run on HPLC for chromatographic separation. Retention time and area under the curve for Mal C (100 μM) and Mal C (100 μM) + NAC (100 μM) are shown in table 1. Overlaid HPLC chromatograms of Mal C and Mal C + NAC are shown in figure 3F. Incubation of Mal C with NAC exhibited no change in retention time (table 1) but significant decrease in the area under curve of Mal C (figure 3F), indicating physical interaction between these two molecules. The experiment was carried out to study the interaction between Mal C and NAC in an aqueous environment by spectral analysis. Mal C (100 μM) was incubated with NAC (100 μM) in water at 37°C, and absorbance spectra were recorded from 250 to 700 nm. Mal C exhibited two absorption maxima at λ1=270 nm and λ2=340 nm. Incubation of Mal C with NAC resulted in a decrease in its absorption peak at λ1=270 nm and an increase in its absorption peak at λ2=340 nm, suggesting interaction between Mal C and NAC (supplementary figure 2).
Mal C treatment depleted cellular thiols significantly at 4 h and 24 h. NAC supplementation enhanced cellular thiols and restored Mal C-mediated depletion of cellular thiols (figure 3G and H).
3.4 Mal C administration inhibited T-cell activation ex vivo
In order to ascertain the ability of Mal C as immunosuppressant in vivo, mice were given intra-peritoneal injection of vehicle (DMSO) or Mal C, and lymphocytes were isolated after 24 h. Then, the cells were stained with CFSE and stimulated with Con A. Mal C administration significantly decreased the percentage of proliferating cells as compared with vehicle control (figure 4A and B). Mal C administration resulted in significant decrease in mitogen-induced cytokine (IL-2, IFN-γ, and IL-6) secretion by T-cells ex vivo (figure 4C).

Mal C treatment suppressed T-cell proliferation and cytokine secretion ex vivo: Mice administered with Mal C (10 mg/kg body weight intraperitoneally) were sacrificed, and the lymphocytes were stained with CFSE followed by stimulation with Con A and cultured for 24 h or 72 h. CFSE fluorescence was monitored by acquiring cells on a flow cytometer. Concatenated flow cytometric histograms are shown (A). The percentage of proliferating cells was calculated based on CFSE dye dilution, and is shown in a bar graph (B). Culture supernatants were harvested from lymphocytes following stimulation with Con A (2.5 µg/mL) after culturing for 24 h and subsequently subjected to cytokine (IL-2, IL-6, and IFN-γ) analysis. The bar graph represents cytokine concentration (pg/mL) (C). Three independent experiments were performed with data points representing means±S.E.M from three replicates from one such experiment (n=3 mice per group). Mal C inhibited GvHD but did not inhibit homeostatic proliferation: Transplantation of allogeneic lymphocytes (10×106 from C57BL/6 mice) into lymphopenic recipients (WBI (6 Gy) BALB/c mice) induced GvHD. The symptoms of GvHD induction such as loss of weight, level of pro-inflammatory cytokines, and survival were monitored. Allogeneic lymphocytes were treated with vehicle or Mal C before transplantation for 4 h, and subsequently, the mice were monitored for 30 days survival and body weight changes. The percentage of survival of mice is represented by a line graph shown in (D), whereas changes in body weight of mice are shown in a line graph in (E). Three independent experiments were performed with 6 mice (n=6) in each group. Data points represent means±SEM from six replicates from 6 mice from one such experiment. To assess the GvHD, mice were sacrificed on day 5 post allogeneic cell transplantation and the levels of serum cytokines were estimated. The concentration of cytokines present in the serum expressed as pg/mL is graphed in (F). Three independent experiments were performed with 3 mice (n=3) in each group. Data points represent mean±SEM from three replicates from one such experiment. For homeostasis-driven proliferation, CD4+ T-cells were isolated from donor BALB/c mice and subsequently stained with CFSE followed by incubation with vehicle or Mal C (10 μM, 4 h). At the end of incubation, 1 million cells were transplanted into syngeneic lymphopenic recipient mice through the tail vein. After 72 h and 96 h, post transplantation, lymphocytes were isolated and subjected to flow cytometric acquiring followed by analysis. Overlaid flow cytometric histograms are shown in (G). The bar graphs show the percentage of proliferating cells in control and Mal C-treated groups at 0, 72, and 96 h, respectively, (H). Three independent experiments were performed with 3 mice (n=3) in each group. Data points represent means±SEM from three replicates from one such experiment.
To investigate the utility of Mal C for prophylaxis of acute GvHD, the lymphocytes from C57BL/6 mice were treated with Mal C (10 μM) for 4 h and used as allogenic donor cells. The recipient BALB/c mice were rendered lymphopenic by exposure to 6 Gy dose of WBI. The donor cells were adoptively transferred by a lateral tail vein injection (10 million cells per mouse) and GvHD was monitored for morbidity and mortality of the hosts. The mice reconstituted with vehicle-treated allogenic cells (GvHD group) died within 30 days. However, Mal C treatment of donor cells completely suppressed GvHD-associated mortality and morbidity (figure 4D and E). GvHD-associated weight loss was evident on day 10 as compared with the unirradiated control, which was significantly prevented in mice reconstituted with Mal C-treated cells (figure 4E). Mice receiving WBI (6 Gy) exhibited significantly elevated IL6 levels as compared with the un-irradiated control (figure 4F). GvHD-associated IFN-γ levels were significantly higher than WBI 6Gy group. Further, serum levels of IL6 and IFN-γ were decreased in hosts reconstituted with Mal C-treated donor cells as compared with the hosts that received vehicle-treated donor cells (figure 4F). Figure 4G shows the effect of Mal C treatment on homeostatic proliferation of syngeneic CD4+ T-cells in lymphopenic hosts. Mal C treatment did not affect the homeostatic proliferation of CD4+ T-cells (figure 4G and H).
4 Discussion
T-cells are important mediators of adaptive immune responses. However, under specific pathological conditions like autoimmunity, allergy, chronic inflammation, COVID-19, and acute GvHD, there is a need to suppress T-cell activation (Maurice et al. 1997; Griffiths et al. 2011; Weyand et al. 2018). The past few decades have seen unprecedented research on the development of prophylactic as well as therapeutic agents to suppress undesirable T-cell activation in the clinic. Many researchers, including our group, have earlier highlighted the influence of cellular redox status and surrounding redox environment in the control of T-cell activation, proliferation, and differentiation (Checker et al. 2009; Kesarwani et al. 2013; Gambhir et al. 2014). Many redox active agents including antioxidants as well as pro-oxidants have been shown to modulate T-cell activation. Previously we demonstrated that pro-oxidants like plumbagin, 1,4-naphthoquinone, and menadione could inhibit T-cell activation at sub-micromolar concentrations through the modulation of GSH levels in cells (Checker et al. 2010, 2011). Antioxidants like GSH, NAC, chlorophyllin, baicalein, and vitamin C have been shown to modulate the activation and differentiation of T-cells (Sharma et al. 2007; Patwardhan et al. 2016). GSH dysregulation is also implicated in early experimental GvHD severity (Suh et al. 2014). Activation of T-cells is also dependent on the intrinsic generation of ROS in the mitochondria and metabolic reprogramming (Sena et al. 2013). Bombay mace, or false nutmeg, is a spice derived from the fruit aril of an Indian medicinal plant Myristica malabarica (Myristicaceae; Ayurvedic name, Rampatri) (Patro et al. 2005). Based on its chemical structure and the known biological properties of its active ingredient, we hypothesized that Mal C may affect T-cell activation. It was indeed found that Mal C treatment significantly suppressed the activation, proliferation, and cytokine secretion in murine T-cells. Further, Mal C treatment suppressed Con A-induced ROS generation. Here we observed that inhibition of T-cell activation was accompanied by a decrease in total ROS levels. Incubation of lymphocytes with Mal C resulted in significant depletion of reduced GSH levels. Hence, a question arises as to whether the efficacy of Mal C as an immune-modulatory agent is due to its antioxidant nature or due to its thiol-seeking behaviour. Interestingly, the anti-proliferative effects of Mal C were abrogated by thiol antioxidants, suggesting possible interaction of Mal C with cellular thiols. Indeed, biophysical studies revealed that Mal C could physically interact with NAC in an aqueous environment. Interestingly, NAC supplementation restored cellular thiol levels in Mal C-treated cells, which could be responsible for the abrogation of anti-proliferative effects of Mal C on T-cells in the presence of thiol antioxidants. These studies revealed that Mal C could possibly interact with cellular protein and non-protein thiols, thereby disrupting cellular redox. Cellular redox changes affect biological responses such as proliferation, differentiation, survival, and apoptosis through redox sensory signaling molecules (Chiu and Dawes 2012; Hancock and Whiteman 2018). MAP kinases can respond to cellular redox changes in terms of their phosphorylation status through the action of redox-sensitive phosphatases (Kamata and Hirata 1999; Seth and Rudolph 2006). T-cell receptor ligation induces distinct MAPK signaling that regulates T-cell responses (Adachi and Davis 2011). Mitogenic stimulation of T-cells leads to phosphorylation of MAP kinases, in turn leading to the activation of immune-regulatory transcription factor NF-κB. Mal C treatment suppressed phosphorylation of ERK and JNK and the subsequent DNA binding of NF-κB following mitogenic stimulation. Upon activation through signals delivered by the T-cell receptor and costimulatory molecules, T-cells upregulate the expression of CD25 (IL2Rα) and CD69 (c-type lectin). While CD25 expression determines IL2 responsiveness, CD69 activation stimulates an influx of calcium ions and the activation of extracellular kinases ERK1/2, thereby facilitating T-cell proliferation (Chen et al. 2020). Mal C-mediated inhibition of both early (CD69) as well as late activation marker (CD25) in T-cells indicates that Mal C interferes with the early activation of T-cells and renders them unresponsive to mitogenic stimulation. Based on these results, we hypothesized that transient exposure of T-cells to Mal C may be beneficial for prophylaxis of acute GvHD. We used a murine model of complete allogenic lymphocyte transplantation and found that Mal C completely abrogated acute GvHD-associated morbidity and mortality of the lymphopenic hosts. Mal C treatment of donor T-cells also resulted in significant reduction in GvHD-associated serum cytokines in the host. In vivo suppression of T-cell proliferation can also result in the disruption of immune homeostasis. In order to study the effect of Mal C on the behaviour of syngeneic T-cells, we evaluated the homeostatic proliferation of purified CD4+ T-cells in syngeneic lymphopenic hosts. Mal C did not inhibit the homeostatic proliferation of T-cells. Mal C treatment led to non-classical cellular redox perturbation by simultaneous scavenging of ROS and thiols. However, glutathione is essential for antigen-induced T-cell proliferation but not for homeostatic proliferation (Sena and Chandel 2012; Sena et al. 2013). Hence, it is anticipated that Mal C treatment did not affect the homeostatic proliferation of T-cells.
The signaling requirements for antigen-specific proliferation of T-cells during acute GvHD are different from those involved in homeostatic proliferation in response to lymphopenia. Our results establish a differential and specific inhibitory action of Mal C on antigen/mitogen-induced proliferation but not on the homeostatic proliferation of T-cells.
In summary, this is the first report showing inhibition of T-cell activation, proliferation, and cytokine production by Mal C through modulation of cellular redox balance (figure 5). Since Mal C suppressed only mitogen/alloantigen-induced proliferation but not homeostatic proliferation, it may be useful for prophylaxis of acute GvHD without disruption of the host immune reconstitution. The present results warrant further mechanistic studies to elucidate the molecular mechanism and biochemical targets of Mal C in T-cells.

Inhibition of T cell responses by Malabaricone C through modulation of cellular redox balance.
Abbreviations
- APC:
-
antigen-presenting cells
- CFSE:
-
carboxyfluorescein diacetate succinimidyl ester
- Con A:
-
concanavalin A
- H2DCFDA:
-
dihydrodichlorofluoroscein diacetate
- DTNB:
-
5,5′-dithio-bis-(2-nitrobenzoic acid)
- ELISA:
-
enzyme-linked immunosorbent assay
- ERK:
-
extracellular related kinase
- FBS:
-
fetal bovine serum
- FITC:
-
fluorescein isothiocyanate
- GSH:
-
glutathione
- GvHD:
-
graft-versus-host disease
- HDP:
-
homeostasis-driven proliferation
- IFN-γ:
-
interferon gamma
- IL2:
-
interleukin 2
- IL6:
-
interleukin-6
- JNK:
-
C-Jun N-terminal kinase
- LPS:
-
lipopolysaccharide
- Mal C:
-
malabaricone C
- NAC:
-
N-acetyl cysteine
- NF-κB:
-
nuclear factor kappa B
- PE:
-
phycoerythrin
- PMSF:
-
phenylmethylsulfonylfluoride
- WBI:
-
whole-body irradiation
References
Adachi K and Davis MM 2011 T-cell receptor ligation induces distinct signaling pathways in naive vs. antigen-experienced T-cells. Proc. Natl. Acad. Sci. USA 108 1549–1554
Banerjee D, Bauri AK, Guha RK, et al. 2008 Healing properties of malabaricone B and malabaricone C, against indomethacin-induced gastric ulceration and mechanism of action. Eur. J. Pharmacol. 578 300–312
Checker R, Sharma D, Sandur SK, et al. 2011 Vitamin K3 suppressed inflammatory and immune responses in a redox-dependent manner. Free Radic. Res. 45 975–985
Checker R, Sharma D, Sandur SK, et al. 2009 Anti-inflammatory effects of plumbagin are mediated by inhibition of NF-kappaB activation in lymphocytes. Int. Immunopharmacol. 9 949–958
Checker R, Sharma D, Sandur SK, et al. 2010 Plumbagin inhibits proliferative and inflammatory responses of T-cells independent of ROS generation but by modulating intracellular thiols. J. Cell. Biochem. 110 1082–1093
Chen Z-Y, Wang L, Gu L, et al. 2020 Decreased expression of CD69 on T-cells in tuberculosis infection resisters. Front. Microbiol. 11 1901
Chiu J and Dawes IW 2012 Redox control of cell proliferation. Trends Cell Biol. 22 592–601
Chong YM, Yin WF, Ho CY, et al. 2011 Malabaricone C from Myristica cinnamomea exhibits anti-quorum sensing activity. J. Nat. Prod. 74 2261–2264
Gagliani N and Huber S 2017 Basic aspects of T helper cell differentiation. Methods Mol. Biol. 1514 19–30
Gambhir L, Checker R, Thoh M, et al. 2014 1, 4-Naphthoquinone, a pro-oxidant, suppresses immune responses via KEAP-1 glutathionylation. Biochem. Pharmacol. 88 95–105
Gelderman KA, Hultqvist M, Holmberg J, et al. 2006 T-cell surface redox levels determine T-cell reactivity and arthritis susceptibility. Proc. Natl. Acad. Sci. USA 103 12831–12836
Gett AV, Sallusto F, Lanzavecchia A, et al. 2003 T-cell fitness determined by signal strength. Nat. Immunol. 4 355–360
Griffiths HR 2005 ROS as signalling molecules in T-cells–evidence for abnormal redox signalling in the autoimmune disease, rheumatoid arthritis. Redox Rep. 10 273–280
Griffiths HR, Dunston CR, Bennett SJ, et al. 2011 Free radicals and redox signalling in T-cells during chronic inflammation and ageing (Portland Press Ltd)
Hancock JT and Whiteman M 2018 Cellular redox environment and its influence on redox signalling molecules. React. Oxyg. Species 5 78–85
Kamata H and Hirata H 1999 Redox regulation of cellular signalling. Cell. Signal. 11 1–14
Kang J, Tae N, Min BS, et al. 2012 Malabaricone C suppresses lipopolysaccharide-induced inflammatory responses via inhibiting ROS-mediated Akt/IKK/NF-κB signaling in murine macrophages. Int. Immunopharmacol. 14 302–310
Kesarwani P, Murali AK, Al -Khami AA,, et al. 2013 Redox regulation of T-cell function: from molecular mechanisms to significance in human health and disease. Antioxid. Redox. Signal. 18 1497–1534
Kundu K and Nayak SK 2017 Total syntheses of malabaricones B and C via a cross-metathesis strategy. J. Nat. Prod. 80 1776–1782
Maurice MM, Nakamura H, Van der Voort E, et al. 1997 Evidence for the role of an altered redox state in hyporesponsiveness of synovial T-cells in rheumatoid arthritis. J. Immunol. 158 1458–1465
Patro BS, Bauri AK, Mishra S, et al. 2005 Antioxidant activity of Myristica malabarica extracts and their constituents. J. Agric. Food Chem. 53 6912–6918
Patro BS, Tyagi M, Saha J, et al. 2010 Comparative nuclease and anti-cancer properties of the naturally occurring malabaricones. Bioorganic Med. Chem. 18 7043–7051
Patwardhan RS, Sharma D, Checker R, et al. 2015 Spatio-temporal changes in glutathione and thioredoxin redox couples during ionizing radiation-induced oxidative stress regulate tumor radio-resistance. Free Radic. Res. 49 1218–1232
Patwardhan RS, Checker R, Sharma D, et al. 2011 Dimethoxycurcumin, a metabolically stable analogue of curcumin, exhibits anti-inflammatory activities in murine and human lymphocytes. Biochem. Pharmacol. 82 642–657
Patwardhan RS, Sharma D, Thoh M, et al. 2016 Baicalein exhibits anti-inflammatory effects via inhibition of NF-κB transactivation. Biochem. Pharmacol. 108 75–89
Sena LA and Chandel NS 2012 Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48 158–167
Sena LA, Li S, Jairaman A, et al. 2013 Mitochondria are required for antigen-specific T-cell activation through reactive oxygen species signaling. Immunity 38 225–236
Seth D and Rudolph J 2006 Redox regulation of MAP kinase phosphatase 3. Biochemistry 45 8476–8487
Sharma D, Eichelberg MR, Haag JD, et al. 2012 Effective flow cytometric phenotyping of cells using minimal amounts of antibody. BioTechniques 53 57–60
Sharma D, Kumar SS and Sainis KB 2007 Antiapoptotic and immunomodulatory effects of chlorophyllin. Mol. Immunol. 44 347–359
Simeoni L and Bogeski I 2015 Redox regulation of T-cell receptor signaling. Biol. Chem. 396 555–569
Suh JH, Kanathezhath B, Shenvi S, et al. 2014 Thiol/redox metabolomic profiling implicates GSH dysregulation in early experimental graft versus host disease (GVHD). PLoS One 9 e88868
Tyagi M, Bhattacharyya R, Bauri AK, et al. 2014a DNA damage dependent activation of checkpoint kinase-1 and mitogen-activated protein kinase-p38 are required in malabaricone C-induced mitochondrial cell death. Biochim. Biophys. Acta 1840 1014–1027
Tyagi M, Patro B and Chattopadhyay S 2014b Mechanism of the malabaricone C-induced toxicity to the MCF-7 cell line. Free Radic. Res. 48 466–477
van Stipdonk MJ, Hardenberg G, Bijker MS, et al. 2003 Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 4 361–365
Weyand CM, Shen Y and Goronzy JJ 2018 Redox-sensitive signaling in inflammatory T-cells and in autoimmune disease. Free Radic. Biol. Med. 125 36–43
Acknowledgements
The authors acknowledge the technical help provided by Deepak V Kathole and BA Naidu during this study.
Funding
To perform studies reported in this work, funds were made available by Department of Atomic Energy, Government of India.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
We confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Additional information
Corresponding editor: Dipankar Nandi
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Patwardhan, R.S., Kundu, K., Purohit, V. et al. Malabaricone C, a constituent of spice Myristica malabarica, exhibits anti-inflammatory effects via modulation of cellular redox. J Biosci 48, 9 (2023). https://doi.org/10.1007/s12038-023-00329-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-023-00329-3