
Abstract
PRNP Q160X is one of the five dominantly inheritable nonsense mutations causing familial prion diseases. Till now, it remains unclear how this type of nonsense mutations causes familial prion diseases with unique clinical and pathological characteristics. Human prion protein (PrP) Q160X mutation is equivalent to Q159X in mouse PrP, which produces the mutant fragment PrP1-158. Through intracerebroventricular injection of recombinant adeno-associated virus in newborn mice, we successfully overexpressed mouse PrP1-158-FLAG in the central nervous system. Interestingly, high level PrP1-158-FLAG expression in the brain caused death in these mice with an average survival time of 60 ± 9.1 days. Toxicity correlated with levels of PrP1-158-FLAG but was independent of endogenous PrP. Histopathological analyses showed microgliosis and astrogliosis in mouse brains expressing PrP1-158-FLAG and most of PrP1-158-FLAG staining appeared intracellular. Biochemical characterization revealed that the majority of PrP1-158-FLAG were insoluble and a significant part of PrP1-158-FLAG appeared to contain an un-cleaved signal peptide that may contribute to its cytoplasmic localization. Importantly, an ~10-kDa proteinase K-resistant PrP fragment was detected, which was the same as those observed in patients suffering from this type of prion diseases. To our knowledge, this is the first animal study of familial prion disease caused by Q159X that recapitulates key features of human disease. It will be a valuable tool for investigating the pathogenic mechanisms underlying familial prion diseases caused by nonsense mutations.





Similar content being viewed by others

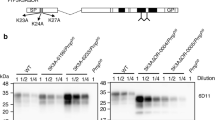
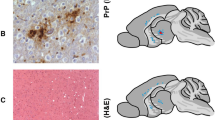
Data Availability
All data are contained within the article. Further inquiries can be directed to the authors.
References
Collinge J (2001) Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 24:519–550. https://doi.org/10.1146/annurev.neuro.24.1.519
Aguzzi A (2006) Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. Journal of neurochemistry 97(6):1726–1739. https://doi.org/10.1111/j.1471-4159.2006.03909.x
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216(4542):136–144. https://doi.org/10.1126/science.6801762
Bolton DC, McKinley MP, Prusiner SB (1982) Identification of a protein that purifies with the scrapie prion. Science 218(4579):1309–1311. https://doi.org/10.1126/science.6815801
Wang F, Wang X, Abskharon R, Ma J (2018) Prion infectivity is encoded exclusively within the structure of proteinase K-resistant fragments of synthetically generated recombinant PrP(Sc). Acta Neuropathol Commun 6(1):30. https://doi.org/10.1186/s40478-018-0534-0
Castle AR, Gill AC (2017) Physiological functions of the cellular prion protein. Front Mol Biosci 4:19. https://doi.org/10.3389/fmolb.2017.00019
Wopfner F, Weidenhofer G, Schneider R, von Brunn A, Gilch S, Schwarz TF, Werner T, Schatzl HM (1999) Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol 289(5):1163–1178. https://doi.org/10.1006/jmbi.1999.2831
Kim MO, Takada LT, Wong K, Forner SA, Geschwind MD (2018) Genetic PrP prion diseases. Cold Spring Harb Perspect Biol 10(5). https://doi.org/10.1101/cshperspect.a033134
Ghetti B, Piccardo P, Zanusso G (2018) Dominantly inherited prion protein cerebral amyloidoses — a modern view of Gerstmann-Straussler-Scheinker. Handb Clin Neurol 153:243–269. https://doi.org/10.1016/B978-0-444-63945-5.00014-3
Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, Kitamoto T, Tateishi J et al (1996) Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci USA 93(2):744–748. https://doi.org/10.1073/pnas.93.2.744
Finckh U, Muller-Thomsen T, Mann U, Eggers C, Marksteiner J, Meins W, Binetti G, Alberici A et al (2000) High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet 66(1):110–117. https://doi.org/10.1086/302702
Jayadev S, Nochlin D, Poorkaj P, Steinbart EJ, Mastrianni JA, Montine TJ, Ghetti B, Schellenberg GD et al (2011) Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol 69(4):712–720. https://doi.org/10.1002/ana.22264
Guerreiro R, Bras J, Wojtas A, Rademakers R, Hardy J, Graff-Radford N (2014) Nonsense mutation in PRNP associated with clinical Alzheimer’s disease. Neurobiol Aging 35 (11):2656.e2613-2656.e2616. https://doi.org/10.1016/j.neurobiolaging.2014.05.013
Fong JC, Rojas JC, Bang J, Legati A, Rankin KP, Forner S, Miller ZA, Karydas AM et al (2017) Genetic prion disease caused by PRNP Q160X mutation presenting with an orbitofrontal syndrome, cyclic diarrhea, and peripheral neuropathy. J Alzheimer’s Dis: JAD 55(1):249–258. https://doi.org/10.3233/JAD-160300
Mead S, Gandhi S, Beck J, Caine D, Gallujipali D, Carswell C, Hyare H, Joiner S et al (2013) A novel prion disease associated with diarrhea and autonomic neuropathy. N Engl J Med 369(20):1904–1914. https://doi.org/10.1056/NEJMoa1214747
Jansen C, Parchi P, Capellari S, Vermeij AJ, Corrado P, Baas F, Strammiello R, van Gool WA et al (2010) Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 119(2):189–197. https://doi.org/10.1007/s00401-009-0609-x
Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L et al (2000) NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 97(1):145–150. https://doi.org/10.1073/pnas.97.1.145
Mohammadi B, Linsenmeier L, Shafiq M, Puig B, Galliciotti G, Giudici C, Willem M, Eden T et al (2020) Transgenic overexpression of the disordered prion protein N1 fragment in mice does not protect against neurodegenerative diseases due to impaired ER translocation. Mol Neurobiol 57(6):2812–2829. https://doi.org/10.1007/s12035-020-01917-2
Miesbauer M, Pfeiffer NV, Rambold AS, Muller V, Kiachopoulos S, Winklhofer KF, Tatzelt J (2009) alpha-Helical domains promote translocation of intrinsically disordered polypeptides into the endoplasmic reticulum. J Biol Chem 284(36):24384–24393. https://doi.org/10.1074/jbc.M109.023135
Heske J, Heller U, Winklhofer KF, Tatzelt J (2004) The C-terminal globular domain of the prion protein is necessary and sufficient for import into the endoplasmic reticulum. J Biol Chem 279(7):5435–5443. https://doi.org/10.1074/jbc.M309570200
Rambold AS, Miesbauer M, Rapaport D, Bartke T, Baier M, Winklhofer KF, Tatzelt J (2006) Association of Bcl-2 with misfolded prion protein is linked to the toxic potential of cytosolic PrP. Mol Biol Cell 17(8):3356–3368. https://doi.org/10.1091/mbc.e06-01-0083
Wang D, Tai PWL, Gao G (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 18(5):358–378. https://doi.org/10.1038/s41573-019-0012-9
Passini MA, Wolfe JH (2001) Widespread gene delivery and structure-specific patterns of expression in the brain after intraventricular injections of neonatal mice with an adeno-associated virus vector. J Virol 75(24):12382–12392. https://doi.org/10.1128/JVI.75.24.12382-12392.2001
Broekman ML, Comer LA, Hyman BT, Sena-Esteves M (2006) Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience 138(2):501–510. https://doi.org/10.1016/j.neuroscience.2005.11.057
Dexter E, Kong Q (2021) Neuroprotective effect and potential of cellular prion protein and its cleavage products for treatment of neurodegenerative disorders part I. a literature review. Expert Rev Neurother 21(9):969–982. https://doi.org/10.1080/14737175.2021.1965881
Matamoros-Angles A, Mohammadi B, Song F, Shafiq M, Brenna S, Puig B, Glatzel M, Altmeppen HC (2023) Inducing prion protein shedding as a neuroprotective and regenerative approach in pathological conditions of the brain: from theory to facts. Neural Regen Res 18(9):1869–1875. https://doi.org/10.4103/1673-5374.366496
Haigh CL, Marom SY, Collins SJ (2010) Copper, endoproteolytic processing of the prion protein and cell signalling. Front Biosci (Landmark Ed) 15(3):1086–1104. https://doi.org/10.2741/3663
Moreno JA, Telling GC (2017) Insights into mechanisms of transmission and pathogenesis from transgenic mouse models of prion diseases. Methods Mol Biol 1658:219–252. https://doi.org/10.1007/978-1-4939-7244-9_16
Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, Jasanoff A, Lindquist S (2009) Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 63(4):438–450. https://doi.org/10.1016/j.neuron.2009.07.026
Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Purro M, Schmid J, Biffiger K et al (2005) Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J 24(13):2472–2480. https://doi.org/10.1038/sj.emboj.7600717
Asante EA, Linehan JM, Tomlinson A, Jakubcova T, Hamdan S, Grimshaw A, Smidak M, Jeelani A et al (2020) Spontaneous generation of prions and transmissible PrP amyloid in a humanised transgenic mouse model of A117V GSS. PLoS Biol 18(6):e3000725. https://doi.org/10.1371/journal.pbio.3000725
Dossena S, Imeri L, Mangieri M, Garofoli A, Ferrari L, Senatore A, Restelli E, Balducci C et al (2008) Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron 60(4):598–609. https://doi.org/10.1016/j.neuron.2008.09.008
Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A et al (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15(6):1255–1264. https://doi.org/10.1002/j.1460-2075.1996.tb00467.x
Telling GC, Tremblay P, Torchia M, Dearmond SJ, Cohen FE, Prusiner SB (1997) N-terminally tagged prion protein supports prion propagation in transgenic mice. Protein Sci 6(4):825–833. https://doi.org/10.1002/pro.5560060409
Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB (1996) Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 10(14):1736–1750. https://doi.org/10.1101/gad.10.14.1736
Piccardo P, Manson JC, King D, Ghetti B, Barron RM (2007) Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci USA 104(11):4712–4717. https://doi.org/10.1073/pnas.0609241104
Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC (1994) Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35(5):513–529. https://doi.org/10.1002/ana.410350504
Masters CL, Gajdusek DC, Gibbs CJ Jr (1981) Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain: a J Neurology 104(3):559–588. https://doi.org/10.1093/brain/104.3.559
Tateishi J, Kitamoto T, Hoque MZ, Furukawa H (1996) Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology 46(2):532–537. https://doi.org/10.1212/wnl.46.2.532
Pirisinu L, Di Bari MA, D’Agostino C, Marcon S, Riccardi G, Poleggi A, Cohen ML, Appleby BS et al (2016) Gerstmann-Straussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep 6:20443. https://doi.org/10.1038/srep20443
Manka SW, Wenborn A, Betts J, Joiner S, Saibil HR, Collinge J, Wadsworth JDF (2023) A structural basis for prion strain diversity. Nat Chem Biol 19(5):607–613. https://doi.org/10.1038/s41589-022-01229-7
Telling GC (2022) The shape of things to come: structural insights into how prion proteins encipher heritable information. Nat Commun 13(1):4003. https://doi.org/10.1038/s41467-022-31460-8
Manka SW, Wenborn A, Collinge J, Wadsworth JDF (2023) Prion strains viewed through the lens of cryo-EM. Cell Tissue Res 392(1):167–178. https://doi.org/10.1007/s00441-022-03676-z
Hallinan GI, Ozcan KA, Hoq MR, Cracco L, Vago FS, Bharath SR, Li D, Jacobsen M (2022) Cryo-EM structures of prion protein filaments from Gerstmann-Straussler-Scheinker disease. Acta Neuropathol 144(3):509–520. https://doi.org/10.1007/s00401-022-02461-0
Ma J, Lindquist S (1999) De novo generation of a PrPSc-like conformation in living cells. Nat Cell Biol 1(6):358–361. https://doi.org/10.1038/14053
Ma J, Lindquist S (2001) Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA 98(26):14955–14960. https://doi.org/10.1073/pnas.011578098
Zanusso G, Petersen RB, Jin T, Jing Y, Kanoush R, Ferrari S, Gambetti P, Singh N (1999) Proteasomal degradation and N-terminal protease resistance of the codon 145 mutant prion protein. J Biol Chem 274(33):23396–23404. https://doi.org/10.1074/jbc.274.33.23396
Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H, Clarke AR, van Leeuwen FW (2007) Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell 26(2):175–188. https://doi.org/10.1016/j.molcel.2007.04.001
Wang X, Wang F, Arterburn L, Wollmann R, Ma J (2006) The interaction between cytoplasmic prion protein and the hydrophobic lipid core of membrane correlates with neurotoxicity. J Biol Chem 281(19):13559–13565. https://doi.org/10.1074/jbc.M512306200
Wu J, Wang X, Lakkaraju A, Sternke-Hoffmann R, Qureshi BM, Aguzzi A, Luo J (2024) Channel activities of the full-length prion and truncated proteins. ACS Chem Neurosci 15(1):98–107. https://doi.org/10.1021/acschemneuro.3c00412
Ma J, Wollmann R, Lindquist S (2002) Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298(5599):1781–1785. https://doi.org/10.1126/science.1073725
Wang X, Bowers SL, Wang F, Pu XA, Nelson RJ, Ma J (2009) Cytoplasmic prion protein induces forebrain neurotoxicity. Biochim Biophys Acta 1792(6):555–563. https://doi.org/10.1016/j.bbadis.2009.02.014
Acknowledgements
We thank Dr Yong-Sun Kim at Ilsong Institute of Life Science, Hallym University, Korea, for the generous gift of 3F10 anti-PrP antibody and Dr Chaoyang Li at Guangzhou Medical University for the generous gift of 8B4 anti-PrP antibody.
Funding
This work was supported by the internal funding from the Chinese Institute for Brain Research, Beijing.
Author information
Authors and Affiliations
Contributions
Conceptualization, J. M.; methodology, Y. Z., R. Y., and J. M.; experimentation, Y. Z., R. Y., and X. Z.; data analysis, Y. Z., R. Y., and J. M.; writing—original draft preparation, J. M., Y. Z.; writing—review and editing, X. Z., Y. Z., R. Y., and J. M.; funding acquisition, J. M.
Corresponding author
Ethics declarations
Ethics Approval
Animal experiments were performed according to the guidelines of the Animal Welfare and Research Ethics Committee of Chinese Institute for Brain Research (No. CIBR-IACUC-039).
Consent to Participate
The manuscript does not contain any data from individual person.
Consent for Publication
All authors approved the publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12035_2024_4224_MOESM1_ESM.docx
Supplementary file1 Supplementary Fig. 1 The expression pattern of PrP1-158-FLAG detected by anti-FLAG antibody differs from that detected by anti-PrP antibodies. A. A schematic diagram illustrating the antigenic epitopes of PrP1-158-FLAG recognized by different PrP antibodies. B. WB analysis with different antibodies to detect the PrP1-158-FLAG expression in the brains injected with rAAV (batch 2).C. WB analysis with 6D11 antibody and anti-FLAG antibody to compare the PrP1-158-FLAG expression pattern in the brains injected with rAAV (batch 1).“WT” and “KO” represent mice with Prnp+/+ and Prnp-/- genotypes, respectively. Supplementary Fig. 2 Verification of the lack of PrP expression in Prnp-/- mice. A. A schematic diagram illustrating the antigenic epitopes of full length PrP recognized by different PrP antibodies. B. WB analysis with different antibodies to detect the expression of PrP in Prnp-/- (KO) and Prnp+/+ (WT) mice. (DOCX 1098 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Y., Yan, R., Zhang, X. et al. Disease-Associated Q159X Mutant Prion Protein Is Sufficient to Cause Fatal Degenerative Disease in Mice. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04224-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04224-2