
Abstract
Understanding the mechanisms underlying amyotrophic lateral sclerosis (ALS) is crucial for the development of new therapies. Previous studies have demonstrated that mitochondrial dysfunction is a key pathogenetic event in ALS. Interestingly, studies in Alzheimer’s disease (AD) post-mortem brain and animal models link alterations in mitochondrial function to interactions between hyperphosphorylated tau and dynamin-related protein 1 (DRP1), the GTPase involved in mitochondrial fission. Recent evidence suggest that tau may be involved in ALS pathogenesis, therefore, we sought to determine whether hyperphosphorylated tau may lead to mitochondrial fragmentation and dysfunction in ALS and whether reducing tau may provide a novel therapeutic approach. Our findings demonstrated that pTau-S396 is mis-localized to synapses in post-mortem motor cortex (mCTX) across ALS subtypes. Additionally, the treatment with ALS synaptoneurosomes (SNs), enriched in pTau-S396, increased oxidative stress, induced mitochondrial fragmentation, and altered mitochondrial connectivity without affecting cell survival in vitro. Furthermore, pTau-S396 interacted with DRP1, and similar to pTau-S396, DRP1 accumulated in SNs across ALS subtypes, suggesting increases in mitochondrial fragmentation in ALS. As previously reported, electron microscopy revealed a significant decrease in mitochondria density and length in ALS mCTX. Lastly, reducing tau levels with QC-01-175, a selective tau degrader, prevented ALS SNs-induced mitochondrial fragmentation and oxidative stress in vitro. Collectively, our findings suggest that increases in pTau-S396 may lead to mitochondrial fragmentation and oxidative stress in ALS and decreasing tau may provide a novel strategy to mitigate mitochondrial dysfunction in ALS.
Graphical abstract

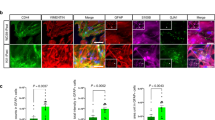
pTau-S396 mis-localizes to synapses in ALS. ALS synaptoneurosomes (SNs), enriched in pTau-S396, increase oxidative stress and induce mitochondrial fragmentation in vitro. pTau-S396 interacts with the pro-fission GTPase DRP1 in ALS. Reducing tau with a selective degrader, QC-01-175, mitigates ALS SNs-induced mitochondrial fragmentation and increases in oxidative stress in vitro.








Similar content being viewed by others


Data Availability
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- AD:
-
Alzheimer’s disease
- DRP1:
-
Dynamin-related protein 1
- SNs:
-
Synaptoneurosomes
- mCTX:
-
Motor cortex
- pTau-S396:
-
Phosphorylated tau at S396
- ROS:
-
Reactive oxygen species
- SOD1:
-
Superoxide dismutase 1
- TARDBP:
-
TAR DNA binding protein
- FUS:
-
Fused in sarcoma
- MAP:
-
Microtubule-associated protein
- OXPHOS:
-
Oxidative phosphorylation
- ALS/FTD:
-
Amyotrophic lateral sclerosis/frontotemporal dementia
- DTT:
-
Dithiothreitol
- SDS:
-
Sodium dodecyl sulphate
- TBST:
-
Tris-buffered saline with Tween 20
- DMEM:
-
Dulbecco’s modified Eagle medium
- FBS:
-
Fetal bovine serum
- ELISA:
-
Enzyme-linked immunosorbent assay
- coIP:
-
Co-immunoprecipitation
- EDTA:
-
Ethylenediaminetetraacetic acid
- PSD-95:
-
Postsynaptic density protein 95
- siRNA :
-
Small Interference RNA
- Mfn1:
-
Mitofusin 1
- Mfn2:
-
Mitofusin 2
- OPA1:
-
Dominant optic atrophy 1
- FDA:
-
Food and Drug Administration
- RNAi:
-
RNA interference
References
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377(2):162–172
Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR, Gitler AD (2020) ALS genetics: gains, losses, and implications for future therapies. Neuron 108(5):822–842
Bodakuntla S, Jijumon AS, Villablanca C, Gonzalez-Billault C, Janke C (2019) Microtubule-associated proteins: structuring the cytoskeleton. Trends Cell Biol 29(10):804–819
Nakamura S, Wate R, Kaneko S, Ito H, Oki M, Tsuge A et al (2014) An autopsy case of sporadic amyotrophic lateral sclerosis associated with the I113T SOD1 mutation. Neuropathology 34(1):58–63
Takeuchi R, Toyoshima Y, Tada M, Tanaka H, Shimizu H, Shiga A et al (2016) Globular glial mixed four repeat Tau and TDP-43 proteinopathy with motor neuron disease and frontotemporal dementia. Brain Pathol 26(1):82–94
Ayaki T, Ito H, Komure O, Kamada M, Nakamura M, Wate R et al (2018) Multiple proteinopathies in familial ALS cases with optineurin mutations. J Neuropathol Exp Neurol 77(2):128–138
Moszczynski AJ, Hintermayer MA, Strong MJ (2018) Phosphorylation of threonine 175 Tau in the induction of Tau pathology in amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD). A review. Front Neurosci 12:259
Stevens CH, Guthrie NJ, van Roijen M, Halliday GM, Ooi L (2019) Increased Tau phosphorylation in motor neurons from clinically pure sporadic amyotrophic lateral sclerosis patients. J Neuropathol Exp Neurol 78(7):605–614
Wang ZX, Tan L, Yu JT (2015) Axonal transport defects in Alzheimer’s disease. Mol Neurobiol 51(3):1309–1321
Stoothoff W, Jones PB, Spires-Jones TL, Joyner D, Chhabra E, Bercury K et al (2009) Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J Neurochem 111(2):417–427
Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI et al (2011) Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol 179(4):2071–2082
Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K et al (2012) Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci 32(7):2430–2441
Rodriguez-Martin T, Cuchillo-Ibanez I, Noble W, Nyenya F, Anderton BH, Hanger DP (2013) Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging 34(9):2146–2157
Cabezas-Opazo FA, Vergara-Pulgar K, Perez MJ, Jara C, Osorio-Fuentealba C, Quintanilla RA (2015) Mitochondrial dysfunction contributes to the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev 2015:509654
Pickett EK, Rose J, McCrory C, McKenzie CA, King D, Smith C et al (2018) Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer's disease. Acta Neuropathol 136(5):747–757
Alavi Naini SM, Soussi-Yanicostas N (2015) Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev 2015:151979
Cheng Y, Bai F (2018) The Association of Tau with mitochondrial dysfunction in Alzheimer’s disease. Front Neurosci. 12:163
Perez MJ, Jara C, Quintanilla RA (2018) Contribution of Tau pathology to mitochondrial impairment in neurodegeneration. Front Neurosci 12:441
Tilokani L, Nagashima S, Paupe V, Prudent J (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 62(3):341–360
Manczak M, Reddy PH (2012) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21(11):2538–2547
Kandimalla R, Manczak M, Fry D, Suneetha Y, Sesaki H, Reddy PH (2016) Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet 25(22):4881–4897
Jiang Z, Wang W, Perry G, Zhu X, Wang X (2015) Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl Neurodegener 4:14
Smith EF, Shaw PJ, De Vos KJ (2019) The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett 710:132933
Calio ML, Henriques E, Siena A, Bertoncini CRA, Gil-Mohapel J, Rosenstock TR (2020) Mitochondrial dysfunction, neurogenesis, and epigenetics: putative implications for amyotrophic lateral sclerosis neurodegeneration and treatment. Front Neurosci 14:679
Obrador E, Salvador R, Lopez-Blanch R, Jihad-Jebbar A, Valles SL, Estrela JM (2020) Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants (Basel) 9(9):901
Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM (1999) Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol 46(5):787–790
Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA (2002) Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem 80(4):616–625
Sasaki S, Iwata M (2007) Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 66(1):10–16
Atsumi T (1981) The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol 55(3):193–198
Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, Feistner H et al (2000) Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 123(Pt 7):1339–1348
Martin LJ (2011) Mitochondrial pathobiology in ALS. J Bioenerg Biomembr 43(6):569–579
Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 181(4):1426–1435
Petrozziello T, Mills AN, Vaine CA, Penney EB, Fernandez-Cerado C, Legarda GPA et al (2020) Neuroinflammation and histone H3 citrullination are increased in X-linked Dystonia Parkinsonism post-mortem prefrontal cortex. Neurobiol Dis 144:105032
Henstridge CM, Sideris DI, Carroll E, Rotariu S, Salomonsson S, Tzioras M et al (2018) Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol 135(2):213–226
Mueller KA, Glajch KE, Huizenga MN, Wilson RA, Granucci EJ, Dios AM et al (2018) Hippo signaling pathway dysregulation in human Huntington’s disease brain and neuronal stem cells. Sci Rep 8(1):11355
Silva MC, Ferguson FM, Cai Q, Donovan KA, Nandi G, Patnaik D et al (2019) Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 8:e45457
Wang P, Geng J, Gao J, Zhao H, Li J, Shi Y et al (2019) Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat Commun 10(1):755
Iijima-Ando K, Sekiya M, Maruko-Otake A, Ohtake Y, Suzuki E, Lu B et al (2012) Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer's disease-related tau phosphorylation via PAR-1. PLoS Genet. 8(8):e1002918
De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF et al (2007) Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet 16(22):2720–2728
Sotelo-Silveira JR, Lepanto P, Elizondo V, Horjales S, Palacios F, Martinez-Palma L et al (2009) Axonal mitochondrial clusters containing mutant SOD1 in transgenic models of ALS. Antioxid Redox Signal 11(7):1535–1545
Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T et al (2011) Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest 121(2):726–738
Janssens J, Wils H, Kleinberger G, Joris G, Cuijt I, Ceuterick-de Groote C et al (2013) Overexpression of ALS-associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol 48(1):22–35
Magrane J, Cortez C, Gan WB, Manfredi G (2014) Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet 23(6):1413–1424
Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW et al (2017) Inhibition of Drp1 ameliorates synaptic depression, Abeta deposition, and cognitive impairment in an Alzheimer’s disease model. J Neurosci 37(20):5099–5110
Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V et al (2017) The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 40(6):583–594 e6
Ruiz A, Quintela-Lopez T, Sanchez-Gomez MV, Gaminde-Blasco A, Alberdi E, Matute C (2020) Mitochondrial division inhibitor 1 disrupts oligodendrocyte Ca(2+) homeostasis and mitochondrial function. Glia 68(9):1743–1756
Hu B, Zhong L, Weng Y, Peng L, Huang Y, Zhao Y et al (2020) Therapeutic siRNA: state of the art. Signal Transduct Target Ther 5(1):101
Scott LJ (2020) Givosiran: First Approval. Drugs 80(3):335–339
Urits I, Swanson D, Swett MC, Patel A, Berardino K, Amgalan A et al (2020) A review of patisiran (ONPATTRO(R)) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther 9(2):301–315
Iwata M, Watanabe S, Yamane A, Miyasaka T, Misonou H (2019) Regulatory mechanisms for the axonal localization of tau protein in neurons. Mol Biol Cell 30(19):2441–2457
Harlen KM, Roush EC, Clayton JE, Martinka S, Hughes TE (2019) Live-cell assays for cell stress responses reveal new patterns of cell signaling caused by mutations in rhodopsin, alpha-synuclein and TDP-43. Front Cell Neurosci 13:535
Xu W, Bao P, Jiang X et al (2019) Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain 142(5):1349–1364
Guerrero EN, Mitra J, Wang H et al (2019) Amyotrophic lateral sclerosis- associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage- mediated neuronal apoptosis. Hum Mol Genet 28(5):2459–2476
Lee A, Rayner SL, Gwee SSL et al (2018) Pathogenic mutation in the ALS/- FTD gene, CCNF, causes elevated Lys48-linked ubiquitylation and defective autophagy. Cell Mol Life Sci 75(2):335–354
Acknowledgments
The authors would like to thank the patients and their families for sample donations.
Funding
T.P. was supported by an award from the Judith and Jean Pape Adams Charitable Foundation and Byrne Family Endowed Fellowship in ALS Research. S.M.K.F. was supported by the ALS Canada Tim E. Noël Postdoctoral Fellowship. S.D. was supported by the Alzheimer’s association (2018-AARF-591935) and the Jack Satter Foundation. D.H.O. is a recipient of an Alzheimer’s Association Clinician Scientist Fellowship (2018-AASCF-592307) and a Jack Satter Foundation Award; he is partially supported by the Dr. and Mrs. E. P. Richardson, Jr Fund for Neuropathology at MGH. S.J.H. was supported by the Alzheimer’s Association/Rainwater Foundation Tau Pipeline Enabling Program and the Stuart & Suzanne Steele MGH Research Scholars Program. The Massachusetts Alzheimer’s Disease Research Center is supported by the National Institute on Aging NIA (Grant P30AG062421). The Philly Dake Electron Microscopy Facility was supported by the Dake Family Foundation and by the NIH grant (1S10RR023594S10) to M.D.
Author information
Authors and Affiliations
Contributions
T.P. and E.A.B. contributed to the study design, data collection, data analysis, and drafting of the manuscript. A.N.M., S.E.K., E.S., B.A.D., A.A.O., S.M.K.F., A.C.A., S.D., and P.M.D. contributed to the data collection, data analysis, and editing of the manuscript. C.H., D.H.O., A.N., B.T.H., T.S.J., S.D.B., K.V., M.E.C., J.D.B., M.D., M.C.S., and S.J.H. contributed to the study design and editing of the manuscript. G.S.V. contributed to the study design, data analysis, and drafting of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
The study was approved by the Mass General Brigham Healthcare Institutional Review Board (IRB).
Consent to Participate
Written informed consent was obtained from all participants prior to study enrollment. Post-mortem consent was obtained from the appropriate representative (next of kin or health care proxy) prior to autopsy.
Consent for Publication
Not applicable.
Conflict of Interest
B.T.H. is a member of Novartis, Dewpoint, and Cell Signaling Scientific Advisory Board (SAB), and of Biogen DMC, and acts as consultant for US DoJ, Takeda, Virgil, W20, and Seer; he receives grants from Abbvie, F prime, NIH, Tau consortium, Cure Alzheimer’s fund, Brightfocus, and JPB foundations. T.S.J. is on the scientific advisory board of Cognition Therapeutics and receives grant funding from European Research Council (grant 681181), UK Dementia Research Institute, MND Scotland, and Autifony. M.E.C. acts as consultant for Aclipse, Mt Pharma, Immunity Pharma Ltd., Orion, Anelixis, Cytokinetics, Biohaven, Wave, Takeda, Avexis, Revelasio, Pontifax, Biogen, Denali, Helixsmith, Sunovian, Disarm, ALS Pharma, RRD, Transposon, and Quralis, and as DSBM Chair for Lilly. J.D.B. has received personal fees from Biogen, Clene Nanomedicine and MT Pharma Holdings of America, and grant support from Alexion, Biogen, MT Pharma of America, Anelixis Therapeutics, Brainstorm Cell Therapeutics, Genentech, nQ Medical, NINDS, Muscular Dystrophy Association, ALS One, Amylyx Therapeutics, ALS Association, and ALS Finding a Cure. S.J.H. is or/has been a member of the SAB and equity holder in Rodin Therapeutics, Psy Therapeutics, Frequency Therapeutics, and Souvien Therapeutics, and has received consulting or speaking fees from Sunovion, Biogen, AstraZeneca, Amgen, Merck, Juvenescence, Regenacy Pharmaceuticals, and Syros Pharmaceuticals, and funding from F-Prime, Tau Consortium, Alzheimer’s Association/Rainwater Foundation Tau Pipeline Enabling Program and the Stuart & Suzanne Steele MGH Research Scholars Program. None of these had any influence over the current paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Supplementary Fig. 1
Antimycin A decrease c- and m-aconitase activity. Antimycin A reduced (a) c-aconitase and (b) m-aconitase activity in SH-SY5Y cells (Mann-Whitney U test=0, p=0.0286, and Mann-Whitney U test=0, p=0.0286, respectively). Data in a-b are represented as bra graph indicating mean ± SD. *p<0.05. (PNG 64 kb)

Supplementary Fig. 2
ALS SNs induce mitochondrial fragmentation. (a) Two-way ANOVA revealed an effect of length ([F(4,234)=1148], p<0.0001) and length X treatment interaction ([F(12,234)=9.629], p<0.0001) in SH-SY5Y cells. Tukey’s test revealed an increase in the frequency of smaller mitochondria (<2μm) in recombinant tau- and ALS SNs-treated cells compared to vehicle- (p<0.0001, and p<0.0001, respectively) and control SNs-treated cells (p<0.0001, and p<0.0001, respectively) as well as a decrease in the frequency of larger mitochondria (>8μm) in recombinant tau- and ALS SNs-treated cells compared to control SNs-treated cells (p=0.0041, and p=0.0003, respectively). Data are represented as scatter plots with bar indicating mean±SD. (b) Two-way ANOVA demonstrated an effect of volume ([F(5,266)=1878], p<0.0001) and volume X treatment interaction ([F(15,266)=5.943], p<0.0001) in SH-SY5Y cells. Tukey’s test revealed an increase in the frequency of smaller mitochondria (<2μm3) in recombinant tau- and ALS SNs- compared to vehicle- (p<0.0001, and p<0.0001, respectively) and control SNs-treated cells (p=0.0028, and p<0.0001, respectively) as well as a decrease in the frequency of larger mitochondria (>10μm3) in ALS SNs- compared to vehicle-treated cells (p=0.0178) and in recombinant tau- and ALS SNs- compared to control SNs-treated cells (p=0.0418, and p<0.0001, respectively). Data are represented as scatter plots with bar indicating mean±SD. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. (PNG 254 kb)

Supplementary Figure 3.
Pro-fusion proteins are not altered in ALS SNs. (a) Representative western blot images of Mfn1, Mfn2 and OPA1 in control and ALS SNs. (b) There was no significant change in Mfn1 (Mann-Whitney U test=144, p=0.8597) (c) Mfn2 (Mann-Whitney U test=61, p=0.9321) or (d) OPA1 levels (Mann-Whitney U test=135, p=0.3994) in ALS SNs (n=32) compared to controls (n=12). Data are represented as individual value plots with the central line representing the median and the whiskers representing the interquartile range. (PNG 264 kb)

Supplementary Figure 4.
siDRP1 decreases DRP1 levels in ALS SNs-treated cells. (a) Representative western blot images of DRP1 in SH-SY5Y cells following siDRP1. DRP1 levels were reduced following transfection with 150nM siDRP1 for 24h and following 48h treatment. (b) There was a significant effect of treatment on DRP1 levels in SH-SY5Y cells (one-way ANOVA, [F(3,12)=18.52], p<0.0001) with a significant decrease in DRP1 levels in siDRP1- and siDRP1+ALS SNs-treated cells compared to siControl- (Tukey’s test, p=0.0023, and p=0.0033, respectively) and siControl+ALS SNs-treated cells (Tukey’s test, p=0.00004, and p=0.0005, respectively). Data are represented as a bar graph with individual values with bar indicating mean ± SD. **p<0.01; ***p<0.001. (PNG 394 kb)

Supplementary Figure 5.
siDRP1 mitigates ALS SNs-induced alteration in mitochondrial connectivity. (a) Representative images of siControl- and siDRP1-transfected SH-SY5Y cells treated with vehicle, recombinant tau, control and ALS SNs stained with Hoechst (blue), CellMask (white), and Tomm20 (red). (b) Cumulative frequency graph indicated an effect of treatment on length (Kruskal-Wallis, H=33.14, p<0.0001) with smaller mitochondria in siControl+tau- and siControl+ALS SNs- (n=3) compared to siControl- (p=0.0023, and p=0.0477, respectively) and siControl+control SNs-treated (n=3) cells (p=0.0008, and p=0.0377, respectively). (c) Cumulative frequency graph indicated an effect of treatment on volume (Kruskal-Wallis, H=40.04, p<0.0001) with smaller mitochondria in siControl+tau- and siControl+ALS SNs- compared to siControl+control SNs-treated cells (p<0.00001, and p<0.0001, respectively). (d) There was no effect of treatment on networks/cell following treatments (two-way ANOVA, [F(3,38)=0.9047], p=0.4479). (e) Two-way ANOVA revealed an effect of treatment ([F(3,39)=3.953], p=0.0149), siDRP1 ([F(1,39)=12.80], p=0.0009), and treatment X siDRP1 interaction ([F(3,39)=5.659], p=0.0026) on large networks/cell in SH-SY5Y cells. Tukey’s test revealed a decrease in tau- and ALS SNs- compared to vehicle- (p=0.0056, and p=0.0374, respectively) or control SNs-treated cells (p=0.0064, and p=0.0416, respectively). siDRP1 prevented tau- and ALS SNs-induced decrease in large networks/cell (Tukey’s test, p=0.0137, and p=0.0082, respectively). (f) Two-way ANOVA demonstrated an effect of treatment ([F(3,39)=4.034], p=0.0136) and treatment X siDRP1 interaction ([F(3,39)=6.527], p=0.0011) on mean branch length. Tukey’s test demonstrated a decrease in siControl+tau and siControl+ALS SNs- compared to siControl- (p=0.0068, and p=0.0195, respectively) or siControl+control SNs-treated cells (p=0.0153, and p=0.0398, respectively). siDRP1 prevented ALS SNs-mediated branch length alterations (p=0.0398). Data in d-f are represented as bar graphs with individual values indicating mean ± SD. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. (PNG 2520 kb)

Supplementary Figure 6.
siDRP1 prevents ALS SNs-induced mitochondrial fragmentation. (a) There was a significant effect of length (two-way ANOVA, [F(4,200)=1502], p<0.0001) and length X treatment interaction (two-way ANOVA, [F(28,200)=3.621], p<0.0001) in SH-SY5Y cells. Tukey’s test revealed a significant increase in the frequency of smaller mitochondria (<2μm) in siControl+tau- and siControl+ALS SNs- (n=3) compared to siControl (p<0.0001, and p=0.0002, respectively) and siControl+control SNs-treated (n=3) cells (p<0.0001 and p=0.0002, respectively). Similarly, Tukey’s test demonstrated a significant decrease in the frequency of larger mitochondria (>8 μm) in recombinant tau- and ALS SNs- compared to siControl-treated cells (p=0.0032 and p=0.0024, respectively) as well as in ALS SNs- compared to control SNs-treated cells (p=0.0433). siDRP1 prevented ALS SNs-induced increases in smaller mitochondria (p=0.0015) and reductions in larger mitochondria (p=0.0433). (b) There was a significant effect of both mitochondrial volume (two-way ANOVA, [F(5,237)=3239], p<0.0001) and volume X treatment interaction (two-way ANOVA, [F(35,237)=3.702], p<0.0001) in SH-SY5Y cells. Tukey’s test revealed a significant increase in the frequency of smaller mitochondria (<2μm3) in siControl+tau- and siControl+ALS SNs compared to siControl (p<0.0001, and p<0.0001, respectively) and control SNs-treated cells (p<0.0001 and p=0.0007, respectively). Similarly, Tukey’s test demonstrated a significant decrease in the frequency of larger mitochondria (>10 μm3) in recombinant tau- and ALS SNs-treated cells compared to siControl (p<0.0001) as well as compared to siControl+control SNs-treated cells (p=0.0203 and p<0.0001, respectively). siDRP1 prevented ALS SNs-induced increases in smaller mitochondria (p=0.0289) and reductions in larger mitochondria (p<0.0001). Data in a-b are represented as bar graphs with individual values indicating mean ± SD. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. (PNG 266 kb)

Supplementary Figure 7.
QC-01-175 prevents ALS SNs-induced mitochondrial fragmentation. (a) There was a significant effect of length (two-way ANOVA, [F(4,215)=955.7], p<0.0001), and length X treatment interaction (two-way ANOVA, [F(28,215)=4.756], p<0.0001). Tukey’s test revealed an increase in the frequency of smaller mitochondria (<2 μm) in recombinant tau- or ALS SNs- (n=3) compared to vehicle- (p=0.0021, and 0.0235, respectively) and control SNs-treated (n=3) cells (p=0.0005, and p=0.0068, respectively). QC-01-175 prevented recombinant tau- and ALS SNs-induced increase in the frequency of smaller mitochondria (p=0.0014 and p<0.0001, respectively). (b) There was a significant effect of volume (two-way ANOVA, [F(5,264)=1510], p<0.0001), and volume X treatment interaction (two-way ANOVA, [F(35,264)=2.864], p<0.0001) in SH-SY5Y cells. Tukey’s test revealed an increase in the frequency of smaller mitochondria (<2 μm3) in recombinant tau- and ALS SNs- compared to vehicle- (p=0.0034, p=0.0027, respectively) and control SNs-treated cells (p=0.0449, and p=0.0373, respectively). QC-01-175 prevented recombinant tau- and ALS SNs-induced increase in the frequency of smaller mitochondria (p=0.0179 and p<0.0001, respectively) as well as ALS SNs-induced decrease in the frequency of larger mitochondria (p=0.0470). Data in a-b are represented as bar graphs with individual values indicating mean ± SD. *p<0.05; **p<0.01; ****p<0.0001. (PNG 262 kb)

Supplementary Figure 8.
Antimycin A decreases ROS levels. Antimycin A decreased ROS levels in SH-SY5Y cells (Mann-Whitney U test=7, p=0.0037). Data are represented as a bar graph indicating mean ± SD. **p<0.01. (PNG 35 kb)
ESM 1
(DOCX 41 kb)
ESM 2
(DOCX 29 kb)
ESM 3
(DOCX 29 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Petrozziello, T., Bordt, E.A., Mills, A.N. et al. Targeting Tau Mitigates Mitochondrial Fragmentation and Oxidative Stress in Amyotrophic Lateral Sclerosis. Mol Neurobiol 59, 683–702 (2022). https://doi.org/10.1007/s12035-021-02557-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02557-w