
Abstract
Severe acute respiratory syndrome coronavirus (SARS-CoV)-2 or COVID-19 has been declared as a pandemic disease by the World Health Organization (WHO). Globally, this disease affected 159 million of the population and reported ~ 3.3 million deaths to the current date (May 2021). There is no definitive treatment strategy that has been identified, although this disease has prevailed in its current form for the past 18 months. The main challenges in the (SARS-CoV)-2 infections are in identifying the heterogeneity in viral strains and the plausible mechanisms of viral infection to human tissues. In parallel to the investigations into the patho-mechanism of SARS-CoV-2 infection, understanding the fundamental processes underlying the clinical manifestations of COVID-19 is very crucial for designing effective therapies. Since neurological symptoms are very apparent in COVID-19 infected patients, here, we tried to emphasize the involvement of redox imbalance and subsequent mitochondrial dysfunction in the progression of the COVID-19 infection. It has been articulated that mitochondrial dysfunction is very apparent and also interlinked to neurological symptoms in COVID-19 infection. Overall, this article provides an in-depth overview of redox imbalance and mitochondrial dysfunction involvement in aggravating COVID-19 infection and its probable contribution to the neurological manifestation of the disease.
Similar content being viewed by others

Introduction
Coronavirus disease 2019 (COVID-19) is the term used by the World Health Organization (WHO) to refer to the unfolding pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It was first described in December 2019 in Wuhan, Hubei Province, China, as pneumonia of unknown cause [1]. Since then, its rapid spread worldwide has become a global health, economic, and humanitarian crisis. The WHO declared COVID-19 a global emergency on January 30, 2020. Subsequently, on March 11, 2020, it was designated as a global pandemic. As of May 2021, it caused over 159 million global cases and ~ 3.3 million deaths in 219 countries and territories around the world (Fig. 1). Although, most people infected with SARS-CoV-2 are either asymptomatic or exhibit mild-to-moderate symptoms, but some of the cases do develop serious and even fatal medical complications. The main clinical neurological manifestations in SARS-CoV-2 infections are fever, dry or productive cough, fatigue, dyspnea, myalgias, headache, sore throat, diarrhea, chills, nausea/vomiting, and rhinorrhea (Fig. 1). However, loss of smell and taste with COVID-19 infection was the most prominent neurological symptom displayed in the patients, and the research has suggested that the inflammation in the olfactory neurons may be linked to this condition. In this review, we have focused on the plausible pharmacological mechanisms associated with the COVID-19 disease progression in perspective of its neurological complications. The incubation period for COVID-19 ranges from 1 to 14 days; however, few cases (less than 1%) have also been reported to develop symptoms after 14 days [2]. Around 75% of cases develop symptoms between 2 and 7 days after exposure with a median incubation period of 4–5 days [3]. Most COVID-19 cases (81%) that experience mild-to-moderate symptoms recover without hospitalization [4] (Fig. 1). Approximately 19% of patients develop severe symptoms and require hospitalization, typically on day 6–8. Few cases (5%) develop critical conditions like respiratory failure, shock, and even multi-organ failure. Generally, the aged population or people with existing chronic medical conditions are at high-risk of developing life-threatening complications with SARS-CoV-2 infection. It is very clear about the incidence rate and patient symptoms of COVID-19 infection, but the uncertainty in the development of this disease challenges us to write this article correlating with existing literature and signifying the importance of fewer molecular targets.

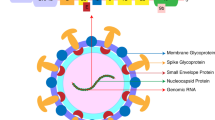
Schematic of the incidence of neurological symptoms and simplified coronavirus replication cycle showing the possible therapeutic targets with potential repurposed drugs. ACE2, angiotensin-converting enzyme carboxypeptidase 2
Structure and Viral Entry into Host
SARS-CoV-2 is a novel coronavirus and is the seventh strain of coronavirus known to infect humans. It is a positive-sense, single-stranded RNA virus and belongs to the genus Betacoronavirus, which also includes SARS-CoV and MERS-CoV [5]. The club-shaped spike projections, originating from the surface of the envelope that contains nucleocapsid, are the most prominent feature of these viruses. The main structural proteins of the coronavirus particles are spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins [5]. The glycoprotein spikes (S protein), present on the surface of the SARS-CoV-2, aid in the binding of the virus to its cellular receptors, the angiotensin-converting enzyme carboxypeptidase 2 (ACE2) (Fig. 1). Subsequently, the conformational changes in the S protein facilitate the internalization, and the ACE2-virus complex is then translocated to endosomes. The fusion of the viral envelope to the cell membrane occurs within acidified endosomes. This ultimately results in the release of viral genomic RNA to the cytoplasm, which is then translated into polypeptides. These polypeptides undergo proteolysis to make nonstructural proteins, some of which form a replication-transcription complex. This complex uses the genomic RNA as a template to replicate newer strands of genomic RNA and to produce subgenomic RNAs by discontinuous transcription on the same template RNA strand. The subgenomic RNAs are translated into structural proteins. The genomic RNA combines with the N protein to form nucleocapsid in the cytoplasm whereas spike (S), envelope (E), and membrane (M) proteins enter the endoplasmic reticulum. Finally, in the endoplasmic reticulum-Golgi intermediate compartment (ERGIC), they assemble with the nucleocapsid to make complete virus particles. Mature virions are released to the extracellular region through exocytosis (Fig. 1). These matured virions further contribute to the cellular damage of the host and thereby exhibits pathogenecity of the infection.
SARS‐CoV‐2: Neurovirulence and Central Nervous System Entry Routes
It was articulated that COVID-19 infection involves a number of neurological symptoms which contribute to morbidity and mortality in infected patients. According to the published reports, the headache, dizziness, impaired consciousness, acute cerebrovascular disease, epilepsy, ataxia, acute disseminated encephalomyelitis (ADEM), viral encephalitis, hyposmia/anosmia, hypogeusia/ageusia, muscle pain, and Guillain–Barre syndrome (GBS) are considered neurological manifestations of COVID-19 infection. The severity of these symptoms is varied depending on overall health status of COVID-19 patients.
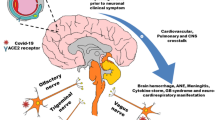
Despite the high prevalence of neurological symptoms and encephalopathy-associated morbidity in COVID-19 patients, the mechanisms underlying these neurological symptoms/manifestations are unclear. SARCoV-2 seems to affect the CNS directly by invading the brain and also indirectly as a consequence of its systemic effects [6] (Fig. 2). The host immune response to the virus itself may affect the CNS by precipitating virus-induced neuropsychiatric sequelae. However, growing evidence is suggesting the neurovirulence of SARS‐CoV‐2 and its role in neuropsychiatric complications [7]. The identification of SARS‐CoV‐2 RNA in the cerebrospinal fluid of COVID-19-associated encephalitis patients provided the most conclusive evidence to document the neurovirulence of SARS‐CoV‐2 [8]. Recently, transmission electron microscopy of the brain tissue of a 74-year-old male with SARS-COV-2 showed evidence indicating the neuroinvasive nature of the virus and the likely routes of transmission to the CNS [9].

SARCoV-2 mediated neurological manifestations. ACE2, angiotensin-converting enzyme carboxypeptidase 2
The key questions for SARS-CoV-2 infection concern the routes of entry into the CNS. One of the probable routes of entry for SARS-CoV-2 into brain tissues could be via the olfactory bulb, which is the only part of the CNS not protected by dura. The presence of anosmia and hyposmia in patients with COVID-19 also supports the idea of SARS-CoV-2 dissemination and spread from the olfactory nerves to the brain [10, 11]. Baig et al. have also suggested that an upper nasal transcribable route enables the SARS‐CoV‐2 to reach the brain [10]. Several studies using experimental animal models have also shown that viruses like human coronavirus OC43 (HCoV-OC43), SARS-CoV-2 and Middle East respiratory syndrome coronavirus (MERS‐COV) could enter the brain following intranasal injection possibly via olfactory nerves with subsequent dissemination to distant area by transneuronal retrograde transport [12,13,14,15]. Similarly, SARS-CoV-2 may also spread to the brain by transneuronal anterograde and retrograde transport via afferent nerve endings of the vagus nerve from lungs [16] and enteric nerve and sympathetic afferents from the gastrointestinal tract [17,18,19]. Moreover, exosomal cellular transport may also be one of the presumed pathways of systemic dissemination of viruses and their subsequent CNS entry [20]. Alternative entry routes include passage across the blood–brain barrier, following viremia, or through infected leukocytes [21,22,23].
The presence of the virus in general circulation enables its entry into cerebral circulation, where slow blood movement in microvessels aids in the binding of viral spike protein with ACE2 receptors present on endothelial cells [10]. This subsequently leads to viral budding from capillary endothelium; resultant damage to the endothelial lining favors viral entry into the milieu of the brain. The angiotensin-converting-enzyme 2 receptor, to which SARS-CoV-2 binds for entry into cells [24], is found in the brain vascular endothelium and smooth muscle [25]. SARS-CoV-2 has been shown to replicate in in vitro neuronal cells [26].
SARS-CoV-2 entry in the brain either via hematogenous route or via neuronal route may lead to cellular dysfunction in the brain cells [27]. ACE2, the functional receptor for SARS-CoV-2, expression in the brain is not only limited to the vasculature but it is also expressed in the neuronal and glial cells [28, 29]. Once in the CNS, it can infect neurons and microglia activating the cascade of neuroinflammation and neurodegeneration through the release of tissue tumor necrosis factor-alpha (TNFα), cytokines, reactive oxygen species (ROS), and other inflammatory mediators. Activated glial cells are well-known to contribute to neuroinflammation by producing pro-inflammatory cytokines like interleukin-6 (IL-6), IL-2, IL-5, and TNFα [30]. Similarly, neuronal production of IL-6 in vivo following viral infection has been reported [31]. SARS-CoV-2 also activates CD4+ cells in the CNS which in turn induces the macrophage to secrete IL-6. IL-6 is a predominant component of the cytokine storm, which is the major cause of COVID-19 associated complications [32]. It is well-established that excessive production of cytokines adversely affects the cellular metabolism in the brain and other organs/tissues. This is further supported by the fact that treatment with tocilizumab (an IL-6 receptor blocker) resulted in the improvement of critically ill patients with COVID-19 [33]. Aforesaid facts suggest that cytokine storm syndrome is one of the main mechanisms associated with SARS-CoV-2-induced brain damage.
SARS-CoV-2 mediated endothelial cell damage of cerebral vasculature and resultant cerebrovascular dysfunction could be another major reason for COVID-19 associated neurological manifestation [6, 27]. Although cerebral vasculature has not yet been investigated; however, endothelial cell infection, inflammation and apoptosis have been seen in various organs including lungs, heart, kidney and bowel at autopsy in patients with COVID-19 [34,35,36].
SARS-CoV-2-induced endothelial cell injury can strongly trigger the coagulation cascade via activation of various pro-coagulation pathways including activation of tissue factor [37, 38]. COVID-19-associated endothelial dysfunction can potentially lead to microvascular and macrovascular complications in the brain [39]. The endothelial damage of cerebral vasculature and subsequent bleeding can have fatal consequences in patients with COVID-19 even without any visible neuronal damage [40]. The persistent inflammatory status in critical patients with COVID-19 patients could also trigger the coagulation cascade. Certain pro-inflammatory cytokines, including IL-6, could activate the coagulation system and suppress the fibrinolytic system. Monocyte activation is postulated to constitute part of the secondary hemophagocytic lymphohistiocytosis described in severe COVID-19 [41]. Thrombocytopenia with elevated d-dimer and C-reactive protein in severe COVID-19 and stroke is consistent with a virus-associated microangiopathic process [42]. Higher production of anti-cardiolipin and anti-β2GP1 antibodies has been also reported in patients with COVID-19 [43]. A recent study has suggested that high morbidity and mortality in COVID-19 patients may be associated with high incidence of acute cerebrovascular disease. This study also suggested that thrombotic microangiopathy in patients with COVID-19 is caused by endotheliopathy with a hemorrhagic predisposition [44]. All these evidences suggest that SARS-CoV-2 mediated endothelial dysfunction of cerebral vasculature and may also contribute to COVID-19-associated neurological manifestations.
Role of COVID-19 in Redox Imbalance and Mitochondrial Dysfunction
Various published literature strongly supports the fact that mitochondrial dysfunction is very apparent in viral infections and may be contributing to immune system dysfunction in the host [45]. However, it is also evident that ACE2 which cleaves Angiotensin II into Angiotensin I to VII plays a vital role in maintaining mitochondrial homeostasis [46] (Fig. 3). An ACE2 knockout mouse has displayed impaired mitochondrial respiration and ATP production [47]. Moreover, NADPH oxidase (Nox4) has been identified as a source of O2− in neuron mitochondria that contributes to ANG II intraneuronal signaling [48]. However, ACE2 importance in COVID-19 entrance into the host immune cells is well-established verity in 2020 [49,50,51]. It is very logical to assume downregulation of ACE2 by COVID-19 may impact on mitochondrial function of immune cells and thereby may reduce the host immune function. Nevertheless, ACE2 has been shown to be under-expressed in experimental models of chronic diseases like diabetes [52], hypertension [53], cardiac myopathy [54], renal dysfunction [55], and neurodegenerative diseases [56]. Our research findings along with others also articulated the significance of mitochondrial dysfunction in the progression of diabetes and its complications: hypertension, Alzheimer’s, Parkinsonism, Huntington’s disease, cardiac myopathy, etc. [57,58,59]. These findings may force us to think that a higher mortality rate is seen in patients with serious COVID-19 infection and suffering from these comorbidities may be associated with mitochondrial dysfunction (Fig. 3). Contrastingly, mitochondrial dysfunction is also found to be independent of the expression of ACE2 in platelets of severely infected COVID-19 patients [60]. This observation states that mitochondrial dysfunction is very significant in COVID-19 disease progression but ACE2 may not be the only for it reason in all infected host cells. Recently, Hoffman et al. demonstrated COVID-19 membrane–associated glycoprotein; Transmembrane serine protease 2 (TMPRSS2) binds with host ACE2, thus helping the virus entry into the host cells [61]. A growing body of evidence suggests that TMPRSS2 also participates in maintaining mitochondrial homeostasis via stimulation of estrogen-related receptor alpha (ERRα)—peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α)—mitochondrial transcription factor (TFAM) axis [62, 63]. PGC1α is considered the master regulator of mitochondrial function and mitochondrial biogenesis [64]. PGC1α knockout mice in different experimental conditions displayed reduced mitochondrial complex activities, ATP production, and dysregulated mitochondrial unfolded response as depicted in Fig. 3 [65, 66]. Based on these observations, there can be a possibility of curtailing COVID-19 infection via boosting PGC1α levels. During COVID-19 infection, it is reported that progressive loss of endothelial barrier in the capillaries supplying the lungs leverages the accumulation of fluid and macromolecules in the interstitium and alveolar spaces which ultimately leads to pulmonary edema formation and in this manner causing severe breathlessness. Adenosine monophosphate–activated protein kinase (AMPK) is considered a metabolic manipulator and critical regulator of endothelial cell function and maintenance of endothelial barrier integrity [67]. A knockout mouse with AMPK has shown significant lung edema, congestion, and inflammation [68]. AMPK activators like AICAR or metformin treatment improved lung function by improving endothelial cells functioning in the blood capillaries supplying blood to the lungs [69, 70]. There are enough literary evidence that has suggested the central role of AMPK in regulating mitochondrial homeostasis [71]. AMPK gets activated in response to the loss of mitochondrial function as an adaptive mechanism; however, during a chronic disease status, AMPK gets compromised where the cellular fate changes with series of pathological events from that of mitochondrial dysfunction to apoptosis [72]. AMPK by activating its downstream signaling proteins maintains mitochondrial function, mitochondrial biogenesis, and mitophagy as shown in Fig. 3. COVID-19 infection downregulates nuclear-encoded mitochondrial genes related to cellular respiration and Complex 1 assembly where they are linked to upstream AMPK activity [73]. Therefore, AMPK activation may be helpful in reducing COVID-19 infection associated with inflammation and mitochondrial dysfunction, and in-depth experimental studies are warranted to understand its role (Fig. 3). Another characteristic feature of COVID-19 infection is the cytokine storm, which is a hyper-inflammatory state of the disease. In patients infected with COVID-19, blood analysis has shown increased TNF-α and inflammatory interleukins which include IL-1β, IL-6, and IL-10 [74]. Several lines of evidence suggested TNFα role in enhancing the mitochondrial ROS generation triggering through calcium-dependent pathways [75]. Additionally, it has also been reported that mitochondrial ROS is one important factor triggering pro-inflammatory cytokines like IL-1β production. IL-1β once induced can lead to inflammasome formation (NLRP3) and may further amplify the persistent inflammation in patients with COVID-19 infection [76]. We also know about the possible involvement of interferon-1 (IFN-1) against viral infections as a part of inbuilt immune defense mechanism [77]. When immune cells are exposed to RNA viruses, an intracellular antiviral response is triggered by activation of RIG-I-like receptors. Thereby, RIG-1-like receptors trigger downstream signaling complex on the mitochondrial outer membrane including the MAVS-TOM70 axis. This can boost the immune response against host viruses via upregulating IFN-β production [78]. Indeed, alternative open reading frame (Orf9) that exists within the nucleocaspid gene of SARS-CoV viruses can significantly inhibit the production of IFN-1 from host mitochondria [79]. Moreover, recent research findings on SARS-CoV-2-infected patients showed increased serum IFN-1 levels. Nevertheless, SARS-CoV-2 Orf9b suppressed type I interferon responses by targeting TOM70 [80]. Altered mitochondrial membrane potential during the viral infections leaks cytochrome c from the mitochondrial matrix into the cytosol [81]. Cytochrome c activates the caspase cascade pathway that leads to programmed cell death or apoptosis [82]. This would be another reason for multiple organ failure–related or breathlessness-related deaths in patients with COVID-19.

Schematic picture showing the plausible mechanism of COVID-19 in mitigating mitochondrial dysfunction. COVID-19 infection stimulates NF-κB pathway and inhibits Nrf2 pathway thereby leads to the redox imbalance and enhances the production of cytokines (TNF-α, IL-1β, IL-6, and IL-10). In turn, TNF-α by acting on its surface receptor enhances the generation of mitochondrial ROS. On another hand, AT1 receptor activation by Ag II formed by ACE involves in regulating AMPK pathway and its downstream mediators controlling mitochondrial function and mitochondrial biogenesis. During the progression of COVID-19 infection, viruses utilize ACE to enter into the host cells and make it unavailable for normal cellular functions, where directly impacts the Ag II homeostasis in controlling cellular functions. This may dysregulate mitochondrial function, perturb mitochondrial biogenesis, and may cause mitochondrial proteotoxicity during COVID-19 infection. Ag II, Angiotensin II; ARE, antioxidant-responsive element; AMPK, adenosine monophosphate–activated protein kinase; Atg, anti-thymocyte globulin; FIP200, FAK family kinase-interacting protein of 200 kDa; GpX, glutathione peroxidase; HIF-1α, hypoxia-inducible factor 1-alpha; HO1, heme oxygenase 1; mTOR, mechanistic target of rapamycin; NQO1, NAD(P)H dehydrogenase [quinone] 1; SIRT1, silent mating–type information regulation 2 homolog 1; NRF, nuclear respiratory factor; Nrf2, nuclear factor erythroid 2 (NFE2)–related factor 2; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; Rheb, Ras homolog enriched in the brain; SOD2, superoxide dismutase 2; TFAM, mitochondrial transcription factor; TSC, tuberous sclerosis proteins; Ulk1, unc-51-like autophagy activating kinase; VPS34, vacuolar protein sorting 34
Mitochondrial ROS directly attack mitochondrial proteins and thus rendering them non-functional. Normally, cells regulate ROS production by enhancing the phase II detoxifying enzymes or also considered antioxidant enzymes; superoxide dismutase (SOD), catalase, NAD(P)H dehydrogenase [quinone] 1 (NQO-1), glutathione peroxidase (GpX), glutathione-S-transferase (GST), and heme oxygenase (HO1) which performs the function of neutralizing the ROS [83]. In this regard, it is very important to discuss nuclear erythroid–related factor 2 (Nrf2) cap ‘n’ collar transcription factor that directly binds to antioxidant responsive elements (ARE) to regulate the gene expression of these antioxidant enzymes [84]. There are several drugs that directly activate Nrf2 under clinical trials for the treatment of more than one indication like sulforaphane and bardoxolone methyl [85, 86]. In previous studies, Nrf2 was under-expressed in human coronavirus HCoV-229E which is associated with the common cold and pulmonary edema [87]. Interestingly, Nrf2 suppression was also found in the lung biopsies of patients with COVID-19 [88]. In fact, ACE2 expression is also regulated by Nrf2 pathway, and few studies demonstrated that Nrf2 under-expression increases the availability of ACE2 and that may increase the risk of viral infection [89]. Mice infected with SARS-CoV have been shown increased NF-κB pathway and produced significant pulmonary inflammation [90]. Many experimental studies advocate a cross-talk between Nrf2 and NF-κB; activation of Nrf2 suppresses the NF-κB activity and subsides over the inflammatory buildup [91, 92]. Additionally, upregulation of HO-1 has been shown to have protective action against many viruses including HIV, hepatitis B and C virus, Enterovirus 71 (E71), influenza virus, respiratory syncytial virus [93], dengue virus (DENV), and Ebola virus (EBOV) [94]. Nrf2 also induces mitochondrial specific antioxidant defense systems like superoxide dismutase-2 (SOD2) which plays a vital role in regulating mitochondrial superoxides [95]. Of note, collective evidence point out Nrf2 would be another potential target for the treatment of COVID-19 infection. Few studies demonstrated that pulmonary complication during COVID-19 infection imparts hypoxia that may cause upregulation of hypoxia-inducible factor-alpha (HIF-1α). HIF-1α downregulates ACE2 levels which may be an adaptive mechanism for the treatment of COVID-19 infection. HIF-1α also have been implicated in regulating heat shock proteins (HSPs) which contribute to repairing misfolded proteins in the mitochondria and impairs mitochondrial dysfunction [96, 97]. Therefore, HIF-1α also may be an important target in manipulating SARS-CoV-2 infection (Fig. 3).
Drugs Under Investigation for COVID-19 Infection Targeting Oxidative Stress, Inflammation, and Mitochondrial Dysfunction
Oxidative stress and inflammation are well-established hallmarks for COVID-19 infection in human beings. Current research in exploring the suitable drug candidates for the treatment of COVID-19 infection is limited with poor availability of experimental models because the SARS-CoV-2 cannot infect wild-type laboratory mice owing to inefficient interactions between the viral spike protein and the mouse orthologue of the human receptor, ACE2. Now, the researchers established a genetic mice model that allows the interaction between SARS-CoV-2 spike proteins and mouse ACE2 and designed mouse-adapted SARS-CoV-2, a recombinant virus that can use mouse ACE2 for entry into cells. This model supported the clinical usage of pegylated IFN-λ1a as a treatment for human COVID-19 infection and under usage. Moreover, several animal models for SARS-CoV-2 have been reported, with varying degrees of viral replication and clinical disease, including ACE2 transgenic mice and virally transduced ACE2 mice, which express human ACE2, ferrets, hamsters, and non-human primates. mRNA vaccines are new type of vaccines which guides the immune cells to synthesize the harmless spike proteins that boosts up the immune system to synthesize antibodies against them and also oppose the actions of actual COVID-19 spike proteins. FDA-approved Moderna and Pfizer/BioNTech’s COVID-19 mRNA vaccine were approved on emergency basis in the USA. The up-to-date research suggests that a prospective drug will be which can affect the viral entrance into the host cells or the drugs previously reported for their antioxidant potentials or drugs used for treatment of inflammatory diseases. Drug repurposing for the treatment/management of COVID-19 infection can lessen the time required for availability of therapy for COVID-19. The summary of the current ongoing trials is summarized in Table 1. These agents are known to target oxidative stress, inflammation, and mitochondrial dysfunction.
Conclusion
This paper reviewed the neurological symptoms associated with COVID-19 infection and presented various mechanisms of SARS-CoV-2 infection related to redox imbalance and mitochondrial dysfunction. Maintaining the redox homeostasis or combating the mitochondrial dysfunction may be as critical as targeting the virus. Treatment strategies that can check the viral entry into the host cell or inhibit the viral infection can be complemented by using strategies like boosting antioxidant defense, improving mitochondrial health and function, and combating the inflammatory surge not only can prevent the viral infection but also can help in reducing the aftermath of viral infection. Further molecular studies deciphering how the agents targeted at redox homeostasis, AMPK modulation, or improving mitochondrial functioning can help in checking the viral entry to host cell are associated tissue/organ damage in COVID-19 infection are warranted.
Data Availability
In this review article, we have not included any data, and all the materials were collected from the published literature resources.
References
Ding Q, Lu P, Fan Y, Xia Y, Liu M (2020) The clinical characteristics of pneumonia patients coinfected with 2019 novel coronavirus and influenza virus in Wuhan. J Med Virol, China. https://doi.org/10.1002/jmv.25781
Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, Azman AS, Reich NG, Lessler J (2020) The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med 172(9):577–582. https://doi.org/10.7326/M20-05042762808
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, Liu L, Shan H et al (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382(18):1708–1720. https://doi.org/10.1056/NEJMoa2002032
Bouadma L, Lescure FX, Lucet JC, Yazdanpanah Y, Timsit JF (2020) Severe SARS-CoV-2 infections: practical considerations and management strategy for intensivists. Intensive Care Med 46(4):579–582. https://doi.org/10.1007/s00134-020-05967-x
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virol J 16(1):69. https://doi.org/10.1186/s12985-019-1182-0
Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, Kneen R, Defres S et al (2020) Neurological associations of COVID-19. Lancet Neurol 19(9):767–783. https://doi.org/10.1016/S1474-4422(20)30221-0
Troyer EA, Kohn JN, Hong S (2020) Are we facing a crashing wave of neuropsychiatric sequelae of COVID-19? Neuropsychiatric symptoms and potential immunologic mechanisms. Brain Behav Immun 87:34–39. https://doi.org/10.1016/j.bbi.2020.04.027
Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, Ueno M, Sakata H et al (2020) A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis 94:55–58. https://doi.org/10.1016/j.ijid.2020.03.062
Paniz-Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J, Sordillo EM, Fowkes M (2020) Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol 92(7):699–702. https://doi.org/10.1002/jmv.25915
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 Virus Targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11(7):995–998. https://doi.org/10.1021/acschemneuro.0c00122
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan. JAMA Neurol, China. https://doi.org/10.1001/jamaneurol.2020.1127.2764549
Glass WG, Subbarao K, Murphy B, Murphy PM (2004) Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. J Immunol 173(6):4030–4039. https://doi.org/10.4049/jimmunol.173.6.4030
Butler N, Pewe L, Trandem K, Perlman S (2006) Murine encephalitis caused by HCoV-OC43, a human coronavirus with broad species specificity, is partly immune-mediated. Virology 347(2):410–421. https://doi.org/10.1016/j.virol.2005.11.044
Tao X, Garron T, Agrawal AS, Algaissi A, Peng BH, Wakamiya M, Chan TS, Lu L et al (2016) Characterization and demonstration of the value of a lethal mouse model of Middle East respiratory syndrome coronavirus infection and disease. J Virol 90(1):57–67. https://doi.org/10.1128/JVI.02009-15
Fenrich M, Mrdenovic S, Balog M, Tomic S, Zjalic M, Roncevic A, Mandic D, Debeljak Z et al (2020) SARS-CoV-2 dissemination through peripheral nerves explains multiple organ injury. Front Cell Neurosci 14:229. https://doi.org/10.3389/fncel.2020.00229
Li YC, Bai WZ, Hashikawa T (2020) The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol 92(6):552–555. https://doi.org/10.1002/jmv.25728
Sanz Segura P, Arguedas Lazaro Y, Mostacero Tapia S, Cabrera Chaves T, Sebastian Domingo JJ (2020) Involvement of the digestive system in covid-19. Gastroenterol Hepatol, A review. https://doi.org/10.1016/j.gastrohep.2020.06.004
Ma C, Cong Y, Zhang H (2020) COVID-19 and the digestive system. Am J Gastroenterol 115(7):1003–1006. https://doi.org/10.14309/ajg.0000000000000691
Wong SH, Lui RN, Sung JJ (2020) Covid-19 and the digestive system. J Gastroenterol Hepatol 35(5):744–748. https://doi.org/10.1111/jgh.15047
Alenquer M, Amorim MJ (2015) Exosome biogenesis, regulation, and function in viral infection. Viruses 7(9):5066–5083. https://doi.org/10.3390/v7092862
Collins AR (2002) In vitro detection of apoptosis in monocytes/macrophages infected with human coronavirus. Clin Diagn Lab Immunol 9(6):1392–1395. https://doi.org/10.1128/cdli.9.6.1392-1395.2002
Desforges M, Le Coupanec A, Dubeau P, Bourgouin A, Lajoie L, Dube M, Talbot PJ (2019) Human coronaviruses and other respiratory viruses: underestimated opportunistic pathogens of the central nervous system? Viruses 12(1):14. https://doi.org/10.3390/v12010014
Cataldi M, Pignataro G, Taglialatela M (2020) Neurobiology of coronaviruses: potential relevance for COVID-19. Neurobiol Dis 143:105007. https://doi.org/10.1016/j.nbd.2020.105007
Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W (2020) Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med 202(5):756–759. https://doi.org/10.1164/rccm.202001-0179LE
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203(2):631–637. https://doi.org/10.1002/path.1570
Chu H, Chan JF, Yuen TT, Shuai H, Yuan S, Wang Y, Hu B, Yip CC et al (2020) Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: an observational study. Lancet Microbe 1(1):e14–e23. https://doi.org/10.1016/S2666-5247(20)30004-5
Alquisiras-Burgos I, Peralta-Arrieta I, Alonso-Palomares LA, Zacapala-Gómez AE, Salmerón-Bárcenas EG, Aguilera P (2020) Neurological complications associated with the blood-brain barrier damage induced by the inflammatory response during SARS-CoV-2 infection. Mol Neurobiol 1–16. https://doi.org/10.1007/s12035-020-02134-7
Turner AJ, Hiscox JA, Hooper NM (2004) ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci 25(6):291–294. https://doi.org/10.1016/j.tips.2004.04.001
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367(6485):1444–1448. https://doi.org/10.1126/science.abb2762
Bohmwald K, Galvez NMS, Rios M, Kalergis AM (2018) Neurologic alterations due to respiratory virus infections. Front Cell Neurosci 12:386. https://doi.org/10.3389/fncel.2018.00386
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S (2008) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol 82(15):7264–7275. https://doi.org/10.1128/JVI.00737-08
Chen C, Zhang XR, Ju ZY, He WF (2020) Advances in the research of mechanism and related immunotherapy on the cytokine storm induced by coronavirus disease 2019. Zhonghua Shao Shang Za Zhi 36(6):471–475. https://doi.org/10.3760/cma.j.cn501120-20200224-00088
Zhang C, Wu Z, Li JW, Zhao H, Wang GQ (2020) Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents 55(5):105954. https://doi.org/10.1016/j.ijantimicag.2020.105954
Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, Bikdeli B, Ahluwalia N et al (2020) Extrapulmonary manifestations of COVID-19. Nat Med 26(7):1017–1032. https://doi.org/10.1038/s41591-020-0968-3
Behzad S, Aghaghazvini L, Radmard AR, Gholamrezanezhad A (2020) Extrapulmonary manifestations of COVID-19: radiologic and clinical overview. Clin Imaging 66:35–41. https://doi.org/10.1016/j.clinimag.2020.05.013
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–1418. https://doi.org/10.1016/S0140-6736(20)30937-5
Bautista-Vargas M, Bonilla-Abadia F, Canas CA (2020) Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. J Thromb Thrombolysis. https://doi.org/10.1007/s11239-020-02172-x
McFadyen JD, Stevens H, Peter K (2020) The emerging threat of (micro)thrombosis in COVID-19 and its therapeutic implications. Circ Res 127(4):571–587. https://doi.org/10.1161/CIRCRESAHA.120.317447
Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J et al (2020) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 191:145–147. https://doi.org/10.1016/j.thromres.2020.04.013
Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, Kucher N, Studt JD et al (2020) Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res 191:9–14. https://doi.org/10.1016/j.thromres.2020.04.024
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395(10229):1033–1034. https://doi.org/10.1016/S0140-6736(20)30628-0
Li Y, Li M, Wang M, Zhou Y, Chang J, Xian Y, Wang D, Mao L et al (2020) Acute cerebrovascular disease following COVID-19: a single center, retrospective, observational study. Stroke Vasc Neurol. https://doi.org/10.1136/svn-2020-000431
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, Chen H, Ding X et al (2020) Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med 382(17):e38. https://doi.org/10.1056/NEJMc2007575
Hernandez-Fernandez F, Valencia HS, Barbella-Aponte RA, Collado-Jimenez R, Ayo-Martin O, Barrena C, Molina-Nuevo JD, Garcia-Garcia J et al (2020) Cerebrovascular disease in patients with COVID-19: neuroimaging, histological and clinical description. Brain. https://doi.org/10.1093/brain/awaa239
Oh H-LJ, Gan SK-E, Bertoletti A, Tan Y-J (2012) Understanding the T cell immune response in SARS coronavirus infection. Emerging Microbes & Infections 1(9):e23
Singh KK, Chaubey G, Chen JY, Suravajhala P (2020) Decoding SARS-CoV-2 hijacking of host mitochondria in pathogenesis of COVID-19. AM J Physiol Cell Physiol. https://doi.org/10.1152/ajpcell.00224.2020
Kim S-M, Kim Y-G, Jeong K-H, Lee S-H, Lee T-W, Ihm C-G, Moon J-Y (2012) Angiotensin II-Induced Mitochondrial Nox4 Is a Major Endogenous Source of Oxidative Stress in Kidney Tubular Cells. PLoS ONE 7(7):e39739
Graham KA, Kulawiec M, Owens KM, Li X, Desouki MM, Chandra D, Singh KK (2010) NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol Ther 10(3):223–231
Dalan R, Bornstein SR, El-Armouche A, Rodionov RN, Markov A, Wielockx B, Beuschlein F, Boehm BO (2020) The ACE-2 in COVID-19: foe or friend? Horm Metab Res 52(5):257
Leng Z, Zhu R, Hou W, Feng Y, Yang Y, Han Q, Shan G, Meng F et al (2020) Transplantation of ACE2-mesenchymal stem cells improves the outcome of patients with COVID-19 pneumonia. Aging Dis 11(2):216
Zangbar HS, Gorji A, Ghadiri T (2020) A review on the neurological manifestations of covid-19 infection: a mechanistic view. Mol Neurobiol 1–14. https://doi.org/10.1007/s12035-020-02149-0
Batlle D, Soler MJ, Ye M (2010) ACE2 and diabetes: ACE of ACEs? Diabetes 59(12):2994–2996
Furuhashi M, Moniwa N, Mita T, Fuseya T, Ishimura S, Ohno K, Shibata S, Tanaka M et al (2015) Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am J Hypertens 28(1):15–21
Bos JM, Hebl VB, Oberg AL, Sun Z, Herman DS, Teekakirikul P, Seidman JG, Seidman CE, Dos Remedios CG, Maleszewski JJ (2020) Marked upregulation of ACE2 in hearts of patients with obstructive hypertrophic cardiomyopathy: implications for SARS-CoV-2-mediated COVID-19. Mayo Clin Pro. https://doi.org/10.1016/j.mayocp.2020.04.028
Reich HN, Oudit GY, Penninger JM, Scholey JW, Herzenberg AM (2008) Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney Int 74(12):1610–1616
Abdelkader NF, Abd El-Latif AM, Khattab MM (2020) Telmisartan/17β-estradiol mitigated cognitive deficit in an ovariectomized rat model of Alzheimer’s disease: modulation of ACE1/ACE2 and AT1/AT2 ratio. Life Sci 245:117388
Beal MF (1998) Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta 1366(12):211–223. https://doi.org/10.1016/s0005-2728(98)00114-5
Kalvala AK, Yerra VG, Sherkhane B, Gundu C, Arruri V, Kumar R, Kumar A Chronic hyperglycemia impairs mitochondrial unfolded protein response and precipitates proteotoxicity in experimental diabetic neuropathy: focus on LonP1 mediated mitochondrial regulation. Pharmacological reports: PR
Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL (2001) Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J Mol Cell Cardiol 33(6):1065–1089
Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben CJ, Petrey AC, Tolley ND, Guo L, Cody MJ (2020) Platelet gene expression and function in COVID-19 patients. Blood. https://doi.org/10.1182/blood.2020007214
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. https://doi.org/10.1016/j.cell.2020.02.052
Xu Z, Wang Y, Xiao ZG, Zou C, Zhang X, Wang Z, Wu D, Yu S et al (2018) Nuclear receptor ERRα and transcription factor ERG form a reciprocal loop in the regulation of TMPRSS2: ERG fusion gene in prostate cancer. Oncogene 37(48):6259–6274
Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A (2004) The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc Natl Acad Sci 101(17):6472–6477
Lynch MR, Tran MT, Parikh SM (2018) PGC1α in the kidney. Am J Physiol-Renal Physiol 314(1):F1–F8
Wan Z, Root-Mccaig J, Castellani L, Kemp BE, Steinberg GR, Wright DC (2014) Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity 22(3):730–738
Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM (2015) Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPRmt. Mol Cell 58(1):123–133
Ayres JS (2020) A metabolic handbook for the COVID-19 pandemic. Nat Metab 1–14. https://doi.org/10.1038/s42255-020-0237-2
Qiu Y-L, Cheng X-N, Bai F, Fang L-Y, Hu H-Z, Sun D-Q (2018) Aucubin protects against lipopolysaccharide-induced acute pulmonary injury through regulating Nrf2 and AMPK pathways. Biomed Pharmacother 106:192–199
Xing J, Wang Q, Coughlan K, Viollet B, Moriasi C, Zou M-H (2013) Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am J Pathol 182(3):1021–1030
Menendez JA (2020) Metformin and SARS-CoV-2: mechanistic lessons on air pollution to weather the cytokine/thrombotic storm in COVID-19. Aging 12(10). https://doi.org/10.18632/aging.103347
Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19(2):121
Carling D (2017) AMPK signalling in health and disease. Curr Opin Cell Biol 45:31–37
Miller B, Silverstein A, Flores M, Xiang W, Cao K, Kumagai H, Mehta HH, Yen K, Kim S-J, Cohen P (2020) SARS-CoV-2 induces a uniquemitochondrial transcriptome signature. Sci Rep. https://doi.org/10.1038/s41598-020-79552-z
Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, Chen L, Li M et al (2020) Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol 11:827
Li X, Fang P, Mai J, Choi ET, Wang H, Yang X-f (2013) Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol 6(1):19
Jo E-K, Kim JK, Shin D-M, Sasakawa C (2016) Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 13(2):148–159
Stetson DB, Medzhitov R (2006) Type I interferons in host defense. Immunity 25(3):373–381
Liu X-Y, Wei B, Shi H-X, Shan Y-F, Wang C (2010) Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res 20(9):994–1011
Shi C-S, Qi H-Y, Boularan C, Huang N-N, Abu-Asab M, Shelhamer JH, Kehrl JH (2014) SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol 193(6):3080–3089
Jiang H-w, Zhang H-n, Meng Q-f, Xie J, Li Y, Chen H, Zheng Y-x, Wang X-n et al (2020) SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol 17(9):998–1000
Lee YJ, Lee C (2018) Porcine deltacoronavirus induces caspase-dependent apoptosis through activation of the cytochrome c-mediated intrinsic mitochondrial pathway. Virus Res 253:112–123
Sahu BD, Kalvala AK, Koneru M, Kumar JM, Kuncha M, Rachamalla SS, Sistla R (2014) Ameliorative effect of fisetin on cisplatin-induced nephrotoxicity in rats via modulation of NF-κB activation and antioxidant defence. PLoS ONE 9(9):e105070
Li W, Kong AN (2009) Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog 48(2):91–104. https://doi.org/10.1002/mc.20465
Yerra VG, Negi G, Sharma SS, Kumar A (2013) Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol 1(1):394–397
Cornblatt BS, Ye L, Dinkova-Kostova AT, Erb M, Fahey JW, Singh NK, Chen M-SA, Stierer T et al (2007) Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis 28(7):1485–1490
Chin MP, Bakris GL, Block GA, Chertow GM, Goldsberry A, Inker LA, Heerspink HJ, O’Grady M et al (2018) Bardoxolone methyl improves kidney function in patients with chronic kidney disease stage 4 and type 2 diabetes: post-hoc analyses from bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes study. Am J Nephrol 47(1):40–47
Gorse GJ, O’Connor TZ, Hall SL, Vitale JN, Nichol KL (2009) Human coronavirus and acute respiratory illness in older adults with chronic obstructive pulmonary disease. J Infect Dis 199(6):847–857
Olagnier DP, Farahani E, Thyrsted J, Cadanet JB, Herengt A, Idorn M, Hait A, Hernaez B, Knudsen A, Iversen MB (2020) Identification of SARSCoV2-mediated suppression of NRF2 signaling reveals a potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat Commun. https://doi.org/10.1038/s41467-020-18764-3
Zhao S, Ghosh A, Lo C-S, Chenier I, Scholey JW, Filep JG, Ingelfinger JR, Zhang S-L et al (2018) Nrf2 deficiency upregulates intrarenal angiotensin-converting enzyme-2 and angiotensin 1–7 receptor expression and attenuates hypertension and nephropathy in diabetic mice. Endocrinology 159(2):836–852
Dosch SF, Mahajan SD, Collins AR (2009) SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-κB pathway in human monocyte macrophages in vitro. Virus Res 142(1–2):19–27
Chen H, Fang Y, Li W, Orlando RC, Shaheen N, Chen XL (2013) NFkB and Nrf2 in esophageal epithelial barrier function. Tissue Barriers 1(5):e27463
Li W, Khor TO, Xu C, Shen G, Jeong W-S, Yu S, Kong A-N (2008) Activation of Nrf2-antioxidant signaling attenuates NFκB-inflammatory response and elicits apoptosis. Biochem Pharmacol 76(11):1485–1489
Brunetti D, Torsvik J, Dallabona C, Teixeira P, Sztromwasser P, Fernandez-Vizarra E, Cerutti R, Reyes A et al (2016) Defective PITRM1 mitochondrial peptidase is associated with Aβ amyloidotic neurodegeneration. EMBO Mol Med 8(3):176–190. https://doi.org/10.1007/s12035-021-02412-y
Espinoza JA, González PA, Kalergis AM (2017) Modulation of antiviral immunity by heme oxygenase-1. Am J Pathol 187(3):487–493
Flynn JM, Melov S (2013) SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radical Biol Med 62:4–12
Genin O, Hasdai A, Shinder D, Pines M (2008) Hypoxia, hypoxia-inducible factor-1α (HIF-1α), and heat-shock proteins in tibial dyschondroplasia. Poult Sci 87(8):1556–1564
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3(3):187–197
Chow JH, Mazzeffi MA, McCurdy MT (2020) Angiotensin II for the treatment of COVID-19–related vasodilatory shock. Anesth Analg. https://doi.org/10.1213/ANE.0000000000004825
Bellucci G, Ballerini C, Mechelli R, Bigi R, Rinaldi V, Reniè R, Buscarinu MC, Baranzini SE, Madireddy L, Matarese G (2020) SARS-CoV-2 metainteractome suggests disease-specific, autoimmune pathophysiologies and therapeutic targets. F1000Research 9:992. https://doi.org/10.12688/f1000research.25593.1
Wu R, Wang L, Kuo H-CD, Shannar A, Peter R, Chou PJ, SLi, R Hudlikar, X Liu, Z Liu, (2020) An update on current therapeutic drugs treating COVID-19. Curr Pharmacol Rep. https://doi.org/10.1007/s40495-020-00216-7
Antwi-Amoabeng D, Kanji Z, Ford B, Beutler BD, Riddle MS, Siddiqui F (2020) Clinical outcomes in COVID-19 patients treated with tocilizumab: an individual patient data systematic review. J Med Virol. https://doi.org/10.1002/jmv.26038
González-García A, García-Sánchez I, Lopes V, Moreno-Arrones OM, Tortosa-Cabañas M, Elías-Sáenz I, Hernández-Rodríguez J (2020) Successful treatment of severe COVID-19 with subcutaneous anakinra as a sole treatment. Rheumatology 59:2171–2173
Sharma S, Ray A, Sadasivam B (2020) Metformin in COVID-19: a possible role beyond diabetes. Diabetes Res Clin Pract 164:108183. https://doi.org/10.1016/j.diabres.2020.108183
Acknowledgements
The authors would like to thank the National Institute of Pharmaceutical Education and Research (NIPER) Kolkata and National Institute of Pharmaceutical Education and Research (NIPER) Raebareli for supporting this work.
Author information
Authors and Affiliations
Contributions
Dr Ravinder K Kaundal (Department of Pharmacology and Toxicology, NIPER-Raebareli, Uttar Pradesh, India—229010) designed the manuscript, prepared figures, and wrote the manuscript; Dr Anil Kumar Kalvala (Department of Pharmaceutical Sciences, Florida Agricultural and Mechanical University, Florida, USA—32301) designed the manuscript, prepared figures, and wrote the manuscript; Dr Ashutosh Kumar (Department of Pharmacology and Toxicology, NIPER-Kolkata, West Bengal, India—700054), supervised, designed manuscript, prepared figures, and wrote manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaundal, R.K., Kalvala, A.K. & Kumar, A. Neurological Implications of COVID-19: Role of Redox Imbalance and Mitochondrial Dysfunction. Mol Neurobiol 58, 4575–4587 (2021). https://doi.org/10.1007/s12035-021-02412-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02412-y