
Abstract
Along with emergence of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in late 2019, a myriad of neurologic symptoms, associated with structural brain changes, were reported. In this paper, we provide evidence to critically discuss the claim that the survived patients could possibly be at increased risk for neurodegenerative diseases via various mechanisms. This virus can directly invade the brain through olfactory bulb, retrograde axonal transport from peripheral nerve endings, or via hematogenous or lymphatic routes. Infection of the neurons along with peripheral leukocytes activation results in pro-inflammatory cytokine increment, rendering the brain to neurodegenerative changes. Also, occupation of the angiotensin-converting enzyme 2 (ACE-2) with the virus may lead to a decline in ACE-2 activity, which acts as a neuroprotective factor. Furthermore, acute respiratory distress syndrome (ARDS) and septicemia induce hypoxemia and hypoperfusion, which are locally exacerbated due to the hypercoagulable state and micro-thrombosis in brain vessels, leading to oxidative stress and neurodegeneration. Common risk factors for COVID-19 and neurodegenerative diseases, such as metabolic risk factors, genetic predispositions, and even gut microbiota dysbiosis, can contribute to higher occurrence of neurodegenerative diseases in COVID-19 survivors. However, it should be considered that severity of the infection, the extent of neurologic symptoms, and the persistence of viral infection consequences are major determinants of this association. Importantly, whether this pandemic will increase the overall incidence of neurodegeneration is not clear, as a high percentage of patients with severe form of COVID-19 might probably not survive enough to develop neurodegenerative diseases.
Similar content being viewed by others

Introduction
In December 2019, a novel coronavirus causing severe acute respiratory syndrome (SARS), shortly named SARS coronavirus 2 (SARS-CoV-2), emerged in China. Until now, this outbreak has been accompanied by a high burden on physical health as well as causing a lot of social, economic and political distress, all over the world [1]. The long-term consequences of the virus, including its effects on physical health, however, might be a way more serious threat to the world. Importantly, this viral infection can cause or even present with neurologic symptoms such as cerebrovascular accident, impaired consciousness, confusion, agitation, seizure, ataxia, headache, anosmia, ageusia, neuropathies, and encephalitis or encephalopathy [2,3,4,5]. In addition, brain structural changes associated with COVID-19 have been confirmed by imaging techniques, both in both surviving patients and non-survivors [6, 7]. Furthermore, based on the evidence for invasion of the virus to the CNS, it has been speculated that different levels of respiratory distress observed in SARS-CoV-2 infection, could be attributed to the effects of the virus on the nervous system [8,9,10]. There is some evidence that human corona viruses can remain latent in the neuronal cell lines [11] and it has been demonstrated that COVID-19 is associated with widespread gene expression changes in the brain, highlighting its long-standing and extensive effects on neurologic function [12]. Thus, apart from the neurologic symptoms in acute phase of infection with the virus, the long-term neurologic sequelae of SARS-CoV-2, for instance neurodegenerative diseases, are of great importance [5].
Neurodegenerative diseases, a group of neurologic diseases characterized by demise of neurons mostly occurring in old age, cause a major burden due to high rates of disability and mortality [13, 14]. A myriad of factors from genetic predispositions to metabolic and environmental risk factors, via various mechanisms including misfolded protein accumulation, impairments in autophagy, mitochondrial dysfunction, neuronal apoptosis, and synaptic impairment, as well as oxidative stress and neuroinflammation contribute to demise of specific neurons in CNS, constituting the pathophysiology of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) [15, 16].
Coronaviruses such as Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV [17] as well as coronavirus disease 2019 (COVID-19) [10] have been shown to cause neurologic disorders after resolution of infection. For instance, it has been determined that viral infections can be associated with some chronic neurological sequelae known as postencephalitic parkinsonism following encephalitis lethargica, which is marked by a set of extrapyramidal symptoms and pathological alterations in substantia nigra and other subcortical nuclei. This entity was first described following influenza pandemics in 1918, but there is no report of such sequelae for human corona virus outbreaks, although there is some indirect evidence for involvement of Corona virus infections in neurodegenerative diseases [18, 19]. Furthermore, human corona virus (HCoV) infection along with other respiratory virus infections such as influenza virus is shown to spread throughout CNS especially temporal region and hippocampus associated with learning/cognitive changes [20]. Whether this hippocampal damage might perpetuate neurodegenerative changes is not clear but AD-specific tau pathology following viral infection-related inflammation associated with cognitive impairments has been confirmed in animal studies [21].
Even ignoring the potential consequences of invasion of the virus to the CNS, COVID-19 is associated with a noticeable increment in inflammatory cytokines which are suggested to lead to a cascade of deleterious mechanisms contributing to neurodegeneration [22]. In addition to direct invasion of the virus to the nervous system and the inflammatory reactions resulting from viral infection, various other mechanisms such as the neuroendocrine axis, cytokine storm, metabolic changes, gut microbiome changes, and hypoperfusion as well as the common underlying risk factors in patients afflicted with SARS-CoV-2 may contribute to the acceleration of neurodegenerative processes. In this article, we aim to revisit the possible pathomechanisms that may contribute to susceptibility to neurodegeneration, especially Alzheimer’s disease and Parkinson’s disease, in patients afflicted with this virus (summarized in Table 1). This might pave the way for designing therapeutic interventions in the future to prevent neurodegeneration in these patients.
Direct or Indirect Penetration to CNS: Neuroinvasive Potential
It has been revealed that SARS-CoV-2 can invade human cells through interaction between the virus S1 spike protein and angiotensin-converting enzyme-2 (ACE-2), which is abundantly expressed in multiple brain tissues [73]. Previously, it was shown that SARS-CoV mRNA, which similarly binds to ACE-2, could be found in brain tissue of the patients infected with this virus [74]. On the whole, the differential levels of respiratory distress, the observed neurologic symptoms in some patients, and structural brain changes in patients have been suggested as clues to the neuroinvasive potential of virus [3, 6,7,8,9,10, 75], although some studies based on CSF analysis, and neuroimaging evidence suggest that neuroinvasion is less likely to be responsible for encephalopathy [37].
Multiple routes have been suggested for SARS-CoV-2 entry into CNS [2, 76]:
-
1)
Interaction of the SARS-CoV-2 with ACE-2 receptor in the capillary endothelium could result in endotheliitis [77] and thereby invasion of the virus into CNS through the destroyed Blood-brain barrier (BBB) could be facilitated. Consistent with this hypothesis, virus RNA and markers of intrathecal inflammation in cerebrospinal fluid (CSF) of patients with COVID-19 infection have been detected [78, 79].
-
2)
Another route proposed for virus entry is hematogenous spread of the virus or its dissemination through lymphatics [80]. Until recently, the commonly held opinion was that brain does not possess characteristic lymphatic drainage, but Loveau et al. (2015) have shown that there exist some lymphatic vessels in the brain which could constitute a proper route for SARS-CoV-2 entry into the brain [23]. In fact, post-mortem studies have revealed that endothelial cells of various tissues could be infected with the virus and the resultant endotheliitis could facilitate virus dissemination through lymphatics and blood vessels resulting in meningitis/encephalitis [77].
-
3)
According to the high rates of anosmia and ageusia observed in patients afflicted with SARS-CoV-2, and its persistence even after recovery from infection in a portion of patients, it has been suggested that SARS-CoV-2 can penetrate nervous system via olfactory bulb, be further disseminated throughout the brain via axonal transport [2, 81]. Additionally, evidence of olfactory bulb asymmetry in a post-mortem brain MRI study on COVID-19 patients was observed [6], although a recent study has suggested that anosmia in COVID-19 patient results from transient edema of olfactory cleft [82]. As ACE-2 and transmembrane serine protease 2 (TRPMSS2), the putative receptors for the virus, are widely expressed on olfactory epithelium a recent study on human olfactory neurons has suggested that virus entry to olfactory neurons occurs via TRPMSS2 receptor, as its expression has been upregulated in olfactory neurons infected with the virus [29]. On the other hand, olfaction dysfunction and structural changes in olfaction-associated areas is an early observation in neurodegenerative diseases such as Parkinson’s disease (PD) and Alzheimer’s disease (AD) [24, 30, 31]. Importantly, persistent hyposmia after recovery from COVID-19 is prevalent among patients with this infection especially in older patients [25]. Accordingly, although the mechanism for post-viral hyposmia is poorly understood [25], but due to expression of both ACE2 and TMPRSS2 in the oligodendrocyte precursor cells and the astrocytes in the substantia nigra, ventricles, and cortex as well as in olfactory bulb pericytes [26,27,28, 83], it has been suggested to be potentially an early sign of Parkinson’s or Alzheimer’s disease [2]. Clinically, this fact might necessitate caution regarding differential diagnosis of anosmia in otherwise asymptomatic patients diagnosed with COVID-19. Mechanistically, the potential of olfactory bulb as a route of virus entry, the probable pathophysiologic changes associated with neurodegenerative diseases, like neuroinflammatory processes or changes in neuronal protein machinery, and the relationship between these conditions should be addressed by further research.
-
4)
Sensory neurons and dorsal root ganglion (DRG) neurons can express ACE-2, as well as other virus receptors such as TMPRSS2 and FURIN [84]. As well, SARS-CoV-2 may find way to the brain through free nerve terminals at the external layers of skin or epithelium of luminal organs [85]. Enteric nervous system and vagus nerve is also considered a potential route of virus entry, compatible with gastrointestinal symptoms observed in patients [86]. Similarly, Parkinson’s disease α-synuclein pathology has been suggested to follow the same pattern of progression, starting from enteric nervous system [87].
Once the virus enters the nervous system, it can bind to the widely expressed ACE-2 in brain tissue and disseminate throughout the brain. As an example, post-mortem electron microscopy performed by Paniz-Monodolfi et al. (2020) on patients with COVID-19 has revealed presence of the virus in the frontal lobe [88]. Importantly, SARS-CoV has been shown to lead to death of neurons in infected mice without causing encephalitis [89]. As well, recently, widespread brain transcriptional changes associated with COVID-19 have been detected [12]. As stated earlier, HCoV infection as well as other respiratory infections, can get disseminated across brain especially temporal and hippocampal regions and there is evidence of AD-specific pathology in hippocampal regions in animal models with viral infections although it is not clear whether it is the result of inflammation or direct viral infiltration [20, 21]. Furthermore, according to Braak hypothesis and staging system in PD, disease-specific pathologic changes in brain regions of patients with PD occur in a characteristic order, starting from enteric nervous system and/or olfactory bulb and then extended through limbic system and cerebral cortices, although this pattern is not universal and there are some controversies over it [87], which is almost consistent with the proposed mechanisms for invasion of the virus.
Cytokine Storm: Neuroimmune Mechanisms
Although there has been a report on SARS-CoV-2 detection in CSF [78], in two reported cases of severe SARS-CoV-2 meningoencephalitis, viral RNA was not detectable, which may be suggestive of transient or undetectable amounts of viral RNA in CNS [90, 91]. On the other hand, cytokine storm, the noticeable increment in circulating levels of cytokines, has been considered one of the main mechanisms responsible for SARS-CoV-2-induced damage to the lungs and death, similar to other members of HCoV family. Upregulation in levels of pro-inflammatory cytokines including interleukin-6 (IL-6) has been detected in patients with COVID-19, especially those with more severe disease [32, 92]. This observation might raise the question that whether it is the peripheral inflammation, rather than direct viral infiltration, that leads to severe meningoencephalitis or other neurologic symptoms. Consistently, high levels of cytokines such as Intelukin-6 (IL-6), IL-8, IL-10, and Tumor Necrosis Factor alpha (TNF-α) as well as positive anti-S1 IgM have been detected in three cases of COVID-19 meningoencephalitis [93]. Indeed, high levels of peripheral cytokines can pass BBB directly and initiate neuroinflammatory reactions or compromise BBB integrity allowing permeation of virus-infected peripheral white blood cells and especially monocytes leading to uncontrolled production of cytokines, microglial activation and neuroinflammation [33, 94]. Immune response dysregulation, on the other hand, is recognized as a main mechanism involved in pathophysiology of neurodegenerative diseases and increment in levels of pro-inflammatory cytokines such as IL-1β and IL-6 is observable in these patients [34, 95]. Although it is not clear whether neuroinflammation can be a trigger for neurodegenerative processes but it has been shown that it can at least accelerate these mechanisms and lead to promotion of neurodegeneration in patients at preclinical stages [95]. Furthermore, it has been revealed that systemic inflammation marked by high cytokine levels can contribute to subsequent hippocampal atrophy, as observed in severe sepsis [96, 97]. Indeed, increased levels of pro-inflammatory cytokines, increase BBB permeability to both cytokines and peripheral leukocytes infected/activated by the virus, which activate resident brain microglia and promote their maturation into the M1 neurotoxic phenotype [40]. This persistent microglial activation can further activate other microglia, promote oxidative stress, propagate tau hyperphosphorylation and protein aggregation, and lead to mitochondrial dysfunction and apoptosis, as well as impairing synaptic plasticity and neurotransmission [41, 98]. Additionally, cytokines resulting from infection, by reaching the brain, can alter synthesis of dopamine, and potentially increase the susceptibility to PD [58]. In cases of SARS-CoV-2, an increased number of CD8 T cells producing interferons including IFN-γ were reported [93], and in SARS-CoV infection, IFN-α and IFN-β have been shown to be effective in limiting virus reproduction [59]. Interferons, like other cytokines, can enhance synaptic loss by activating microglia [99] and in the presence of β-amyloids, these peptides act as part of host immune system and can entrap viral particles, promoting the post-viral inflammatory response induced by interferons [35].
The upregulation in cytokine levels can be, at least partly, attributed to Corona virus open reading frame 3a (ORF3a) protein and aggravated by the associated Acute Lung Injury (ALI), which are recognized to induce NOD-like receptor protein 3 (NLRP3) inflammasome activity and promote cytokine production, namely IL-1β [36, 100]. On the other hand, systemic inflammation, through activation of NLRP3 inflammasome, impairs immune homeostasis in the brain, promotes production of pro-inflammatory cytokines particularly IL-1β and propagates aggregation of peptides in form of pathogenic fibrils, as well as causing mitochondrial dysfunction and apoptosis thereby contributing to neurodegeneration, as in AD and PD [42, 43, 71]. All such evidence highlights the fact that NLRP3 inflammasome might act as the cross-road in potential neurodegenerative consequences of SARS-CoV-2 infection.
An important question that will be raised is whether SARS-CoV-2 infection has long-standing effects and whether the resultant cytokine storm will persist and can potentially lead to neuropathologic changes associated with neurodegenerative diseases. Actually, answer to this question might depend on multiple factors such as the extent to which there is an underlying ground for inflammation, the extent of potential neuroinvasion or migration of peripheral leukocytes to the brain, or the presence of autoimmune reactions due to mechanisms such as molecular mimicry following the viral infection. In this regard, COVID-19 has been known to cause autoimmune reactions by molecular mimicry mechanisms causing cranial neuropathies [85], multiple sclerosis [101], Guillain–Barré syndrome [10], or encephalomyeloradiculitis [72]. In keeping with this, higher levels of anti-corona virus antibodies have been detected in CSF sample of PD patients [102] and it has been determined that specific variants of the virus such as mutations in glycoprotein (S) by inducing immune reactions can precipitate glutamate excitotoxicity and thereby contribute to neurodegeneration [103]. Furthermore, it has been determined that under inflammatory conditions especially in old ages, BBB integrity is compromised and myeloid cells infected with the virus can migrate to CNS and perpetuate neuroinflammation by cytokine production and prompting microglial activation [104, 105]. Over time, these leukocytes may remain infected with the HCoV and act as a persistent source of inflammation in the brain, contributing to the neurodegenerative milieu [106].
Adoptive and Cellular Immune System: Immune Cells in Brain
The role of cellular immune system in COVID-19 infection has been highlighted in some recent studies. Actually, it has been revealed that SARS corona virus structural and non-structural antigens activate CD4 and CD8 T cell response, occurring proportionate to the severity of infection [107, 108]. In mild cases, the CD8 T cell response against virus is dominant, but in severe cases, the humoral immune response and CD4 T cells take over the antiviral immune response more prominently, which might itself even contribute to immunopathology [108]. Furthermore, the systemic inflammatory response induced by viral infection could alter the anti-inflammatory conditions in favor of inflammatory conditions [108]. Whether or not the cellular and adoptive immune system activation in periphery influence central nervous system is not clear, but it has been revealed that in viral infections, due the BBB breakdown, white blood cells could further penetrate BBB and perpetuate infection in brain and culminate in neuroinflammatory processes [76]. In COVID-19 infection, elevations in cellular markers of inflammation, such as increased white blood cells, were not observed in CSF samples in a study with a small sample size [79], although in some cases, pleocytosis in CSF samples has been reported [37, 38]. In post-mortem histopathological analysis of patients who died of COVID-19 infection, severe microglial activation and mild perivascular infiltration of T cells was noted in CNS [39, 45]. This might suggest that although prominent immune cell activation is not common in mild and moderate infections, in severe forms of the disease, cellular components of immune system infiltrate in CNS. As mentioned earlier, infiltration of microglia in CNS is associated with dysregulated production of cytokines, disruption of neurotransmission, and even hippocampal atrophy [97] as well as propagation of neurodegenerative mechanisms and apoptosis [41, 98]. Furthermore, evidence T cell infiltration in the CNS has been detected in both AD [44] and PD [109]. This inflammatory, cytotoxic polarization of immune cells would more possibly occur in the old patients with COVID-19 infection [110] and further propagate the neurodegenerative mechanisms, which may have already been triggered by misfolded protein accumulations due to aging [57]. On the whole, due to the variable cellular immune response in different phases of infection, and in mild or severe forms of infection [108], the effects of such changes on neurodegenerative processes, especially in the long run, cannot be clearly predicted.
Interactions of Viral Proteins: Protein Aggregation and Mitochondrial Dysfunction
Infection of dopaminergic neurons expressing α-synuclein with respiratory viruses such as H1N1 has culminated in α-synuclein aggregate formation and suppression of autophagy, the main mechanisms involved in priming neurodegeneration in PD [111]. On the other hand, SARS-CoV-2 proteins can interact with human proteins, including proteins associated with aging such as those related to ubiquitination, mitochondrial activity, RNA processing, or vesicle trafficking [55]. It was determined that SARS-CoV infection can hijack endoplasmic reticulum (ER) machinery for synthesis of viral proteins, and its spike protein by interacting with ER chaperons activates PERK pathway which in turn stimulates glucose-regulated protein 78 (GRP78), and GRP94, inducing the unfolded protein response (UPR), thereby enhancing protein misfolding [56]. On the other hand, Open Reading Frame 9b (ORF-9b) of SARS-CoV can induce autophagy of the host cell, as well as inducing ubiquitination, and impairing proteostasis in mitochondria [112]. Furthermore, SARS-CoV proteins, ORF-3a, ORF-3b, ORF-6, ORF-7a, can induce apoptosis of host cells, which is mediated by caspase-3 and ER stress and Janus Kinase (JNK) pathways [113]. Although such evidence is not available for SARS-CoV-2 yet, but the structural and mechanistic behavior similarity of these viruses makes these assumptions so likely that targeting ER stress has been suggested as a treatment for SARS-CoV-2 [114, 115]. Thus, it is possible that COVID-19 infection, enters the neurons, by hijacking protein machinery disrupts ER and mitochondrial function and upregulates accumulation of misfolded proteins, thereby triggers mitochondrial oxidative stress and contributes to apoptosis and degeneration of neurons [55, 60, 116].
Gut Microbiome: Shared Mechanisms of Immune System and Gastrointestinal Tract
According to a recent systematic review and meta-analysis study, around 7.8% and 5.5% of patients with COVID-19 infection experience gastrointestinal symptoms such as diarrhea and nausea/vomiting, respectively, as well as other symptoms like abdominal pain and GI bleeding. This indicates virus infiltration in the GI epithelium, possibly through ACE receptors, although the mechanism is not well determined [52, 117]. Importantly, shedding of the virus in feces of afflicted patients has been observed in five weeks or more after infection [53]. Furthermore, various genetic and environmental factors including infections are key to the composition of gut microbiota and such alterations have been detected in COVID-19 patients [54, 118]. On the other hand, as mentioned earlier, it is mechanistically possible that pathogens find their way into CNS via peripheral nerve endings and contribute to neurodegeneration and gut microbiota dysbiosis can potentially play a prominent role in dissemination of the virus and its invasion into CNS. Furthermore, based on the findings in animal models and humans, it has been speculated that some alterations in GI tract, e.g. GI lesions and increased permeability of intestinal epithelium, occurs decades before development of neurodegenerative disease-specific pathologies and might contribute to development of neurodegenerative diseases [119].
Besides the possible contribution of gut microbiota dysbiosis to direct invasion of the virus into CNS, by mechanisms like increasing GI permeability, molecular mimicry, and oxidative stress upregulates immune system activation, which along with altering neurotransmission balance it can contribute to neurodegenerative processes [119]. Having such facts in mind, it might be possible that SARS-CoV-2 infection, by modifying gut microbiota renders the patients to developing neurodegenerative diseases [119]. However, it is not clear whether any potential alterations in composition of gut microbiome would be permanent and whether it depends on the severity of infection and the presence of gastrointestinal symptoms. In this regard, the recent systematic review on GI manifestations of COVID-19 has shown that there is no significant difference in prevalence of GI symptoms in severe and non-severe forms of the disease [52].
Furthermore, gut microbiota composition plays an important role in maintaining the immunological equilibrium and is suggested as a determinant for rates of affliction with infectious and inflammatory diseases including COVID-19 [48, 120]. For instance, old age, as a common risk factor for severe form of COVID-19 infection and neurodegenerative diseases is associated with less variety in gut microbiota [120]. This fact might indicate that since the diversity of gut microbiota acts as a common risk factor for COVID-19 infection and development of neurodegeneration, the odds for developing neurodegenerative diseases would be higher in patients surviving COVID-19.
Considering all of mentioned links between gut microbiome, COVID-19 infection and neurodegeneration, will illuminate that personalized nutritional modifications and maybe fecal transplantation methods might have a preventive role for COVID-19 infection and even be beneficial in diminishing the risk of neurodegeneration in patients with history of COVID-19 infection [120].
Angiotensin-Converting Enzymes: Neuroprotective and Neurotoxic Features
Angiotensin-converting enzymes (ACE), consisting of ACE-1 and ACE-2, are key components of renin-angiotensin system (RAS). ACE-2 is a membrane-bound enzyme expressed on many cell types, which is responsible for cleavage of angiotensin II into smaller proteins such as angiotensin (1-7) which bind to Mas receptor, forming ACE-2/angiotensin (1-7)/Mas axis [49]. As mentioned earlier, ACE-2 acts as a receptor which binds to SARS-CoV-2 spike protein, thereby allowing its entry to cells [51]. Binding of SARS-CoV-2 to ACE-2 can potentially lead to ACE-2 depletion, which can further perpetuate multiple organ injury, as ACE-2/angiotensin (1-7)/Mas axis is known to have protective effects [50]. On the other hand, similar protective role of ACE-2/angiotensin (1-7)/Mas axis in neurodegeneration has been addressed, contrary to ACE-1 and angiotensin II which are shown to be involved in augmentation of oxidative stress, neuroinflammation, and apoptosis, thereby contributing to neurodegeneration [121]. Lower levels of ACE-2 and angiotensin (1-7) activity, and higher levels of ACE-1 activity and angiotensin II, in association with higher levels of Aβ and phosphorylated tau, in AD patients compared to controls [68], as well as similar evidence in animal AD models [69] has also been detected.
Apart from the role of ACE-2 and its downstream axis in demise of neurons, gene expression studies have revealed that ACE-2 gene shows the most significant co-expression with dopamine decarboxylase, which is responsible for conversion of L-3,4-dihydroxyphenylalanine (L-DOPA) into dopamine. This indicates that ACE-2 downregulation, induced by SARS-CoV-2 infection, might be associated with concomitant alterations in dopamine synthetic pathway, which is implicated in pathophysiology of PD [122]. Furthermore, ACE-2 gene polymorphisms have been shown to influence susceptibility to infection with COVID-19 and its complications such as multi-organ failure, although the exact association has not been determined [123, 124], while specific ACE-2 polymorphisms can potentially increase the risk for PD [125] and AD [70].
Accordingly, ACE inhibitors by reducing angiotensin II levels have shown promise in preventing progression of neurodegenerative diseases [121, 126]. Then again the question that will be raised is whether such alterations in ACE-2 levels will persist or not. Moreover, whether or not ACE inhibitor administration would halt perpetuation of organ injury and the potential for progression to neurodegeneration in patients with severe COVID-19 infection remains a question.
Hypothalamic-Pituitary Axis: Neuroendocrine Mechanisms
The over-activation of immune system and cytokine production in acute phase of viral infections is speculated to stimulate hypothalamic-pituitary-adrenocortical (HPA) axis [68, 69]. This phenomenon will lead to secretion of higher amounts of glucocorticoids, which downregulate activation of inflammatory cells and production of excessive amounts of cytokines, in order to prevent the deleterious effects of cytokine storm [122]. In addition to the increased production of glucocorticoids as a physiologic response to the viral infection, administration of exogenous glucocorticoids in the critically ill patients with COVID-19 might further add to the increased levels of glucocorticoids in these patients. Importantly, prolonged amounts of glucocorticoids are known to accelerate neurodegenerative processes through a myriad of mechanisms. Disturbances in glucocorticoids can lead to mitochondrial dysfunction and enhance apoptosis, activate hyperphosphorylation of tau, the key cytoskeleton protein, and thereby contribute to synaptic loss and impaired neurotransmission [127]. Consistent with this, in patients with de novo PD, HPA axis abnormality is observed as a common finding [128].
On the other hand, SARS-CoV can express certain sequences of amino acid which are structurally similar to adrenocorticotropic hormone (ACTH), and the host immune response by forming antibodies, can destroy the endogenous ACTH [129]. Moreover, it is suggested that SARS-CoV infection, reduces ACE-2 expression in various tissues such as lung and heart [130, 131]. On the other hand, ACE-2 overexpression in corticotropin-releasing hormone (CRH) producing cells in hypothalamus is associated with a decrement in HPA axis activation [132]. It is not clear whether SARS-CoV-2 infection reduces ACE-2 expression in these hypothalamic neurons [133], but if so, this mechanism will further lead to HPA axis activation [132]. Despite the changes in HPA axis described in acute phase of viral infections, 3 months after recovery from SARS-CoV a subset of patients showed HPA axis hypo-activity without any abnormalities in HPA axis prior to or during viral infection [134]. This phenomenon can be attributed to some cytokines such as transforming growth factor-beta (TGF-β) and TNF-α, which are suggested to downregulate activation of HPA axis under certain circumstances and are shown to be involved in severe SARS-CoV-2 infection [135, 136]. However, additional data is required to determine whether the effects of COVID-19 infection on HPA axis function is similar to SARS-CoV, whether TGF-β mediates such effects and what would be the long-term consequences of this infection on HPA axis, if any.
Hypoxia and Thrombosis: Systemic or Local Insults to the Brain
Acute respiratory distress syndrome (ARDS), considered the most serious clinical presentation of COVID-19 [137], especially when requiring long-term mechanical ventilation, is accompanied by higher rates of subsequent cognitive impairment and executive dysfunction in survivors, even years after hospital discharge [138]. The risk for developing dementia and/or progressive neurodegeneration in patients with ARDS depends on multiple factors like pre-existing neurologic injury, dementia, amyloid-beta pathology or delirium, and presence of concomitant hypoperfusion or hypoxemia, cytokine levels, and incompetent BBB integrity [138]. Actually, ARDS-related hypoxemia and systemic inflammation/sepsis as well as increased BBB permeability are the main mechanisms known to contribute to brain damage. Consistent with this, a recent post-mortem study on COVID-19 patients has shown neuropathological changes consistent with hypoxic injury in majority of patients, despite minimal SARS-CoV-2 infection in brain tissue [5]. Also recently, it has been revealed that in patients even with moderate SARS-CoV-2 infection, the levels of glial fibrillary acidic protein (GFAP) and neurofilament light chain protein (NFL) are elevated, which are indicators of astrocytic reaction and axonal injury, respectively [139]. Nevertheless, it has been determined that hippocampal damage, specifically in CA1 and CA2, is attributable more to high levels of cytokines rather than hypoxemia [140]. Indeed, upregulation in levels of peripheral cytokines and sepsis is associated with alterations in function of epithelial cells in BBB and subsequent tendency towards amyloid-beta peptide build-up and AD pathology [141]. As stated earlier, sepsis can also trigger neuroinflammatory processes in the brain, alter glucose metabolism and neurotransmission, and increase brain tissue vulnerability to systemic insults such as oxidative stress [142]. As well, septic shock, especially when associated with hypercapnia, can impair auto-regulatory mechanisms for cerebral blood circulation [143], further contributing to hypoxemia. Apart from ARDS and the subsequent blood circulation alterations, local vascular obstruction in CNS may be precipitated by sepsis-induced coagulopathy, which is the tendency towards formation of micro-thrombosis in end-organs induced by systemic inflammation and is prominent in critically ill COVID-19 patients, causing defects in brain circulation and render patients to hypoxic-ischemic changes [144, 145]. Furthermore, SARS-CoV-2 by binding to ACE-2 can potentially cause ACE-2 depletion and by shifting the equilibrium towards ACE1/angiotensin II, the subsequent vasoconstriction can further contribute to hypoperfusion [144]. As mentioned in the previous section, ACE-2 cleaves angiotensin II to angiotensin (1-7), which as well as inducing vasodilation, by binding to Mas receptor exerts anti-inflammatory effects and is recognized as a neuroprotective factor [146]. Importantly, it has become evident that Alzheimer-type dementia and other neurodegenerative diseases may be precipitated by chronic cerebral hypoperfusion and vascular event [46]. Moreover, thrombosis and endothelial dysfunction are associated with impaired anti-oxidant activity, which along with hypoxia results in oxidative stress and production of inflammatory cytokines, the main contributors to neurodegeneration [47]. Oxidative stress can enhance α-synuclein aggregation in PD [147], beta-amyloid peptide accumulation in AD [62] through neuroinflammation and other mechanisms.
APOE and Lipid Metabolism: Shared Genetic and Metabolic Risk Factors
Delirium and impaired consciousness is a common clinical finding, especially in severe cases of COVID-19 [75] and pre-existing dementia has been recognized to increase the risk for severity of COVID-19 infection about three-fold as well as increasing mortality [63]. Moreover, it has been determined that a large proportion of COVID-19 reported symptoms are inheritable, especially delirium was reported to have 50% heritability, according to the study in homozygotic twins [61]. Interestingly, ApoE e4-e4 genotype is associated with a significantly increased risk for dementia and delirium [64] and it has been shown that ApoE e4 homozygotes, independent from pre-existing dementia or other comorbidities, were more probably tested positive for COVID-19 and would probably experience more severe forms of the disease [66, 148]. This same genetic predisposition increasing the risk for AD and COVID-19, might constitute a confounding factor which at least partly explains the possibly higher risks of developing AD-type dementia in patients surviving COVID-19.
Mechanistically, it has been shown that high levels of blood cholesterol by binding to ApoE receptor, improve SARS-CoV-2 entry to cells via ACE-2 receptors [149]. ApoE e4 genotype, on the other hand, favors pro-inflammatory conditions in macrophages, and has lower efficiency in delivering essential fatty acids for maintenance of neuronal membrane and myelin sheath, as well as promoting misfolded protein accumulations, disrupting dendritic spine formation and synaptic plasticity [150].
In one study from China on patients surviving SARS-CoV, it was shown that 12 years following recovery from infection, patients developed metabolic changes especially lipid metabolism disruptions such as increase in levels of phosphatidylinositol and lysophosphatidylinositol and hyperlipidemia, associated with hyperinsulinemia, impaired glucose metabolism or cardiovascular abnormalities [151]. Such metabolic changes were suggested to be associated with high-dose pulses of methylprednisolone, as well as the severity of initial damage to the lungs [151]. Also, binding of the virus to cells led to acute T2DM during SARS-CoV infection in 50% of patients without history of Type 2 diabetes mellitus (T2DM), although only 5% of them remained diabetic after resolution of infection, with no significant difference in blood glucose levels compared to their siblings [67]. These permanent metabolic changes such as DM were especially observed in critically ill patients with complications after discharge from hospital, which might occur due to pancreatic damage or consequences of septicemia and the associated metabolic changes [65]. Additionally, type 1 diabetes mellitus (T1DM) due to autoimmune mechanisms or damage to pancreatic islet cells following viral infection is a phenomenon that might occur in these patients [67]. Even more, long-term cognitive dysfunction [138], as well as depression and post-traumatic stress disorder (PTSD) [152] following ARDS and hypoxia might be associated with poor episodic and working memory and uncontrolled eating behaviors, culminating in weight gain [153]. Although occurrence of such metabolic changes, following SARS-CoV-2 infection is not established yet, but should be anticipated. Importantly, obesity, metabolic syndrome, lipid metabolism alterations and diabetes mellitus are known to increase the odds for affliction with neurodegenerative disorders [154].
Also, increment in blood glucose levels in T2DM patients may aggravate fibril formation and AD pathology, through upregulation of interferon regulatory factor 5 (IRF5) activity [35]. Increased IRF-5 in response to high blood glucose levels, on the other hand, promotes susceptibility for COVID-19 infection [35]. Moreover, it has been recently shown that obesity and metabolic syndrome increase the susceptibility for affliction with this infection [155]. Thus, even if we assume that rate of metabolic changes following COVID-19 infection will not increase significantly, we can consider the fact that patients with pre-existing metabolic abnormalities are recognized to be at higher risk of COVID-19 and more severe form of the disease [156], and the same population, if surviving COVID-19, are at greater risk for developing neurodegenerative disorders [154].
Conclusion
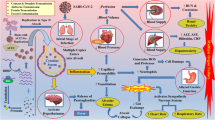
As discussed throughout this article and summarized in Table 1, Fig. 1, and Fig. 2, direct or indirect invasion of the SARS-CoV-2 into CNS and migration and infiltration of peripheral leukocytes to the brain induces production of cytokines, activates microglia, induces their polarization into M1 phenotype, and thereby contributes to the propagation of neurodegenerative processes. Viral replication can also hijack protein synthesis machinery in neurons, promote unfolded protein response, and by impairing proteostasis trigger misfolded protein accumulation. On the other hand, sepsis and elevated cytokine production will upregulate HPA axis and cortisol secretion, which can further enhance tau hyperphosphorylation. ACE-2 depletion due to its excessive occupation of with SARS-CoV-2, and the subsequent decline in ACE-2/angiotensin (1-7)/Mas axis activity will attenuate neuroprotective mechanisms and promote neurodegeneration. Additionally, ARDS as well as sepsis-induced hypercoagulation render brain to hypoxic damage, which is known to cause oxidative stress and enhance neurodegenerative processes. The shared risk factors for developing neurodegenerative diseases and infection with COVID-19 including genetic variances such as APOE e4-e4 genotype, and metabolic risk factors such as Diabetes Mellitus or hyperlipidemia, could also underlie the higher risk for developing neurodegenerative diseases in recovered patients, if any.

Contribution of SARS-CoV-2 to neurodegeneration via invasion into CNS. SARS-CoV-2 can directly invade olfactory bulb, penetrate brain via retrograde axonal transport from peripheral nerve endings, or reach brain via hematogenous pathway. Virus invasion along with excessive peripheral cytokine production, due to the septicemia, upregulate central cytokine production and will enhance the microglial activation, which along with T cell infiltration lead to neuroinflammation and contribute to neurodegeneration. Furthermore, it has been shown that SARS-CoV spike protein can promote Endoplasmic reticulum (ER) unfolded protein response, impair autophagy and proteostasis in mitochondria and culminate in misfolded protein accumulation and apoptosis

Contribution of SARS-CoV-2 to neurodegeneration via its systemic effects. Occupation of the angiotensin-converting enzyme-2 (ACE-2) by the virus will reduce ACE-2/Angiotensin (1-7)/Mas axis activity, which acts as a neuroprotective mechanism. Acute respiratory distress syndrome (ARDS)-induced hypoxemia along with the sepsis-induced hyper-coagulation and the resultant tendency towards formation of local thrombosis in brain vessels will cause hypoperfusion exacerbate oxidative stress and promote neurodegeneration
Whether or not such mechanisms will persist and the extent to which they can accelerate the process of neurodegeneration in patients with variable disease severity is not clear. Additionally, due to the high rates of death, especially in severe forms of the disease and in patients with comorbidities which increase the risk for neurodegeneration, as well as premature death in these patients due to other comorbidities, it is possible that the overall prevalence/incidence of neurodegenerative diseases will not increase in the upcoming years. On the whole, in this time the research on the association between these entities is based on hypothesis and for providing evidence in this regard, prospective cohort studies should be designed. Furthermore, it is noteworthy that the increased risk for development of neurodegenerative diseases in this population is not inevitable; for instance, modifying neurodegenerative mechanisms by Vitamin D supplementation has been suggested [157]. Similarly, targeting ACE and RAS system by administering ACE inhibitors may be a promising method to halt neurodegeneration.
Change history
27 January 2021
A Correction to this paper has been published: https://doi.org/10.1007/s12035-021-02308-x
References
Nicola M, Alsafi Z, Sohrabi C, Kerwan A, Al-Jabir A, Iosifidis C, Agha M, Agha R (2020) The socio-economic implications of the coronavirus pandemic (COVID-19): a review. Int J Surg 78:185–193. https://doi.org/10.1016/j.ijsu.2020.04.018
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 77(6):683–690. https://doi.org/10.1001/jamaneurol.2020.1127
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, Collange O, Boulay C et al (2020) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med 382(23):2268–2270. https://doi.org/10.1056/NEJMc2008597
Montalvan V, Lee J, Bueso T, De Toledo J, Rivas K (2020) Neurological manifestations of COVID-19 and other coronavirus infections: a systematic review. Clin Neurol Neurosurg 194:105921. https://doi.org/10.1016/j.clineuro.2020.105921
Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, Kneen R, Defres S et al Neurological associations of COVID-19. Lancet Neurol. https://doi.org/10.1016/S1474-4422(20)30221-0
Coolen T, Lolli V, Sadeghi N, Rovaï A, Trotta N, Taccone FS, Creteur J, Henrard S et al (2020) Early postmortem brain MRI findings in COVID-19 non-survivors. Neurology. https://doi.org/10.1212/wnl.0000000000010116
Chougar L, Shor N, Weiss N, Galanaud D, Leclercq D, Mathon B, Belkacem S, Stroër S et al (2020) Retrospective observational study of brain magnetic resonance imaging findings in patients with acute SARS-CoV-2 infection and neurological manifestations. Radiology 2020:202422. https://doi.org/10.1148/radiol.2020202422
Gandhi S, Srivastava AK, Ray U, Tripathi PP (2020) Is the collapse of the respiratory center in the brain responsible for respiratory breakdown in COVID-19 patients? ACS Chem Neurosci 11(10):1379–1381. https://doi.org/10.1021/acschemneuro.0c00217
Nouri-Vaskeh M, Sharifi A, Khalili N, Zand R, Sharifi A (2020) Dyspneic and non-dyspneic (silent) hypoxemia in COVID-19: possible neurological mechanism. Clin Neurol Neurosurg 198:106217. https://doi.org/10.1016/j.clineuro.2020.106217
Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, Franciotta D, Baldanti F et al (2020) Guillain–Barré syndrome associated with SARS-CoV-2. N Engl J Med 382(26):2574–2576. https://doi.org/10.1056/NEJMc2009191
Arbour N, Côté G, Lachance C, Tardieu M, Cashman NR, Talbot PJ (1999) Acute and persistent infection of human neural cell lines by human coronavirus OC43. J Virol 73(4):3338–3350. https://doi.org/10.1128/jvi.73.4.3338-3350.1999
Yang AC, Kern F, Losada PM, Maat CA, Schmartz G, Fehlmann T, Schaum N, Lee DP, Calcuttawala K, Vest RT, Gate D, Berdnik D, McNerney MW, Channappa D, Cobos I, Ludwig N, Schulz-Schaeffer WJ, Keller A, Wyss-Coray T (2020) Broad transcriptional dysregulation of brain and choroid plexus cell types with COVID-19. bioRxiv:2020.2010.2022.349415. doi:https://doi.org/10.1101/2020.10.22.349415
Dorsey ER, Elbaz A, Nichols E, Abd-Allah F, Abdelalim A, Adsuar JC, Ansha MG, Brayne C et al (2018) Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17(11):939–953. https://doi.org/10.1016/S1474-4422(18)30295-3
Nichols E, Szoeke CEI, Vollset SE, Abbasi N, Abd-Allah F, Abdela J, Aichour MTE, Akinyemi RO et al (2019) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18(1):88–106. https://doi.org/10.1016/S1474-4422(18)30403-4
Castillo X, Castro-Obregón S, Gutiérrez-Becker B, Gutiérrez-Ospina G, Karalis N, Khalil AA, Lopez-Noguerola JS, Rodríguez LL et al (2019) Re-thinking the etiological framework of neurodegeneration. Front Neurosci 13:728. https://doi.org/10.3389/fnins.2019.00728
Gitler AD, Dhillon P, Shorter J (2017) Neurodegenerative disease: models, mechanisms, and a new hope. Disease Models &. Mechanisms 10(5):499–502. https://doi.org/10.1242/dmm.030205
Kim JE, Heo JH, Kim HO, Song SH, Park SS, Park TH, Ahn JY, Kim MK et al (2017) Neurological complications during treatment of Middle East respiratory syndrome. J Clin Neurol 13(3):227–233. https://doi.org/10.3988/jcn.2017.13.3.227
Cheyette SR, Cummings JL (1995) Encephalitis lethargica: lessons for contemporary neuropsychiatry. J Neuropsychiatr Clin Neurosci 7(2):125–134. https://doi.org/10.1176/jnp.7.2.125
Giordano A, Schwarz G, Cacciaguerra L, Esposito F, Filippi M (2020) COVID-19: can we learn from encephalitis lethargica? Lancet Neurol 19(7):570. https://doi.org/10.1016/s1474-4422(20)30189-7
Hosseini S, Wilk E, Michaelsen-Preusse K, Gerhauser I, Baumgärtner W, Geffers R, Schughart K, Korte M (2018) Long-term neuroinflammation induced by influenza A virus infection and the impact on hippocampal neuron morphology and function. J Neurosci 38(12):3060–3080. https://doi.org/10.1523/jneurosci.1740-17.2018
Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM (2011) Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol 178(6):2811–2822. https://doi.org/10.1016/j.ajpath.2011.02.012
Serrano-Castro PJ, Estivill-Torrús G, Cabezudo-García P, Reyes-Bueno JA, Ciano Petersen N, Aguilar-Castillo MJ, Suárez-Pérez J, Jiménez-Hernández MD et al (2020) Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: a delayed pandemic? Neurología (English Edition) 35(4):245–251. https://doi.org/10.1016/j.nrleng.2020.04.002
Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, Herod SG, Knopp J et al (2018) CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci 21(10):1380–1391. https://doi.org/10.1038/s41593-018-0227-9
Silva MME, Mercer PBS, Witt MCZ, Pessoa RR (2018) Olfactory dysfunction in Alzheimer’s disease systematic review and meta-analysis. Dement Neuropsychol 12(2):123–132. https://doi.org/10.1590/1980-57642018dn12-020004
Lee J, Nallani R, Cass L, Bhalla V, Chiu A, Villwock J (2020) A systematic review of the neuropathologic findings of post-viral olfactory dysfunction: implications and novel insight for the COVID-19 pandemic. Am J Rhinol Allergy 1945892420957853. doi:https://doi.org/10.1177/1945892420957853
Guo A-X, Cui J-J, OuYang Q-Y, He L, Guo C-X, Yin J-Y (2020) The clinical characteristics and mortal causes analysis of COVID-19 death patients. medRxiv:2020.2004.2012.20062380. doi:https://doi.org/10.1101/2020.04.12.20062380
Qi J, Zhou Y, Hua J, Zhang L, Bian J, Liu B, Zhao Z, Jin S (2020) The scRNA-seq expression profiling of the receptor ACE2 and the cellular protease TMPRSS2 reveals human organs susceptible to COVID-19 infection. bioRxiv. https://doi.org/10.1101/2020.04.16.045690
Brann D, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, Chance R, Macaulay I et al (2020) Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. bioRxiv. https://doi.org/10.1101/2020.03.25.009084
Bagasra O, Pandey P, McCean E, Albrecht H Infectivity of human olfactory neurons to SARS-CoV-2: a link to anosmia.
Zapiec B, Dieriks BV, Tan S, Faull RLM, Mombaerts P, Curtis MA (2017) A ventral glomerular deficit in Parkinson’s disease revealed by whole olfactory bulb reconstruction. Brain 140(10):2722–2736. https://doi.org/10.1093/brain/awx208
Sanjari Moghaddam H, Dolatshahi M, Salardini E, Aarabi MH (2019) Association of olfaction dysfunction with brain microstructure in prodromal Parkinson disease. Neurol Sci 40(2):283–291. https://doi.org/10.1007/s10072-018-3629-2
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395(10229):1033–1034. https://doi.org/10.1016/s0140-6736(20)30628-0
Mohammadi S, Moosaie F, Aarabi MH (2020) Understanding the immunologic characteristics of neurologic manifestations of SARS-CoV-2 and potential immunological mechanisms. Mol Neurobiol 57(12):5263–5275. https://doi.org/10.1007/s12035-020-02094-y
Ng A, Tam WW, Zhang MW, Ho CS, Husain SF, McIntyre RS, Ho RC (2018) IL-1β, IL-6, TNF-α and CRP in elderly patients with depression or Alzheimer’s disease: systematic review and meta-analysis. Sci Rep 8(1):12050. https://doi.org/10.1038/s41598-018-30487-6
Naughton SX, Raval U, Pasinetti GM (2020) Potential novel role of COVID-19 in Alzheimer’s disease and preventative mitigation strategies. J Alzheimers Dis 76:21–25. https://doi.org/10.3233/JAD-200537
Siu K-L, Yuen K-S, Castano-Rodriguez C, Ye Z-W, Yeung M-L, Fung S-Y, Yuan S, Chan C-P et al (2019) Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J 33(8):8865–8877. https://doi.org/10.1096/fj.201802418R
Tuma R, Guedes B, Carra R, Iepsen B, Rodrigues J, Camelo Filho AE, Kubota G, Ferrari M, Studart-Neto A, Oku M, Terrim S, Lopes C, Passos Neto CE, Dalben M, De Souza J, Baima JP, Da Silva T, Perissinotti I, Martin MDG, Goncalves M, Fortini I, Smid J, Adoni T, Lucatto L, Nitrini R, Gomes H, Castro LH (2020) Clinical, cerebrospinal fluid and neuroimaging findings in COVID-19 encephalopathy: a case series. medRxiv:2020.2008.2028.20181883. doi:https://doi.org/10.1101/2020.08.28.20181883
de Oliveira FAA, Palmeira DCC, Rocha-Filho PAS (2020) Headache and pleocytosis in CSF associated with COVID-19: case report. Neurol Sci 41(11):3021–3022. https://doi.org/10.1007/s10072-020-04694-x
Hanley B, Naresh KN, Roufosse C, Nicholson AG, Weir J, Cooke GS, Thursz M, Manousou P et al (2020) Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: a post-mortem study. Lancet Microbe 1(6):e245–e253. https://doi.org/10.1016/s2666-5247(20)30115-4
Grozdanov V, Bliederhaeuser C, Ruf WP, Roth V, Fundel-Clemens K, Zondler L, Brenner D, Martin-Villalba A et al (2014) Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol 128(5):651–663. https://doi.org/10.1007/s00401-014-1345-4
Sawada M, Imamura K, Nagatsu T (2006) Role of cytokines in inflammatory process in Parkinson’s disease. J Neural Transm Suppl 70:373–381. https://doi.org/10.1007/978-3-211-45295-0_57
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S et al (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575(7784):669–673. https://doi.org/10.1038/s41586-019-1769-z
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D et al (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493(7434):674–678. https://doi.org/10.1038/nature11729
Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, Oda T, Tsuchiya K et al (2002) Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol 124(1-2):83–92. https://doi.org/10.1016/s0165-5728(01)00496-9
Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, Kneen R, Defres S et al (2020) Neurological associations of COVID-19. Lancet Neurol 19(9):767–783. https://doi.org/10.1016/S1474-4422(20)30221-0
Park J-H, Hong J-H, Lee S-W, Ji HD, Jung J-A, Yoon K-W, Lee J-I, Won KS et al (2019) The effect of chronic cerebral hypoperfusion on the pathology of Alzheimer’s disease: a positron emission tomography study in rats. Sci Rep 9(1):14102. https://doi.org/10.1038/s41598-019-50681-4
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12(12):723–738. https://doi.org/10.1038/nrn3114
van der Lelie D, Taghavi S (2020) COVID-19 and the gut microbiome: more than a gut feeling. mSystems 5(4):e00453–e00420. https://doi.org/10.1128/mSystems.00453-20
Tikellis C, Thomas MC (2012) Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept 2012:256294. https://doi.org/10.1155/2012/256294
Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, Hou C, Wang H et al (2020) Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care 24(1):422. https://doi.org/10.1186/s13054-020-03120-0
South AM, Brady TM, Flynn JT (2020) ACE2 (angiotensin-converting enzyme 2), COVID-19, and ACE inhibitor and Ang II (angiotensin II) receptor blocker use during the pandemic. Hypertension 76(1):16–22. https://doi.org/10.1161/HYPERTENSIONAHA.120.15291
Suresh Kumar VC, Mukherjee S, Harne PS, Subedi A, Ganapathy MK, Patthipati VS, Sapkota B (2020) Novelty in the gut: a systematic review and meta-analysis of the gastrointestinal manifestations of COVID-19. BMJ Open Gastroenterol 7(1):e000417. https://doi.org/10.1136/bmjgast-2020-000417
Parasa S, Desai M, Thoguluva Chandrasekar V, Patel HK, Kennedy KF, Roesch T, Spadaccini M, Colombo M et al (2020) Prevalence of gastrointestinal symptoms and fecal viral shedding in patients with coronavirus disease 2019: a systematic review and meta-analysis. JAMA Netw Open 3(6):e2011335. https://doi.org/10.1001/jamanetworkopen.2020.11335
Gu S, Chen Y, Wu Z, Chen Y, Gao H, Lv L, Guo F, Zhang X et al (2020) Alterations of the gut microbiota in patients with COVID-19 or H1N1 influenza. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa709
Lippi A, Domingues R, Setz C, Outeiro TF, Krisko A (2020) SARS-CoV-2: at the crossroad between aging and neurodegeneration. Mov Disord 35(5):716–720. https://doi.org/10.1002/mds.28084
Chan C-P, Siu K-L, Chin K-T, Yuen K-Y, Zheng B, Jin D-Y (2006) Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol 80(18):9279–9287. https://doi.org/10.1128/jvi.00659-06
Krisko A, Radman M (2019) Protein damage, ageing and age-related diseases. Open Biol 9(3):180249. https://doi.org/10.1098/rsob.180249
Miller AH, Haroon E, Raison CL, Felger JC (2013) Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety 30(4):297–306. https://doi.org/10.1002/da.22084
Sainz B Jr, Mossel EC, Peters CJ, Garry RF (2004) Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV). Virology 329(1):11–17. https://doi.org/10.1016/j.virol.2004.08.011
Scheper W, Hoozemans JJM (2015) The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol 130(3):315–331. https://doi.org/10.1007/s00401-015-1462-8
Williams FM, Freydin M, Mangino M, Couvreur S, Visconti A, Bowyer RC, Le Roy CI, Falchi M, Sudre C, Davies R, Hammond C, Menni C, Steves C, Spector T (2020) Self-reported symptoms of COVID-19 including symptoms most predictive of SARS-CoV-2 infection, are heritable. medRxiv 2020.2004.2022.20072124. doi:https://doi.org/10.1101/2020.04.22.20072124
Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F (2018) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol 14:450–464. https://doi.org/10.1016/j.redox.2017.10.014
Atkins JL, Masoli JA, Delgado J, Pilling LC, Kuo C-LC, Kuchel G, Melzer D (2020) Preexisting comorbidities predicting severe COVID-19 in older adults in the UK Biobank Community Cohort. medRxiv 2020.2005.2006.20092700. doi:https://doi.org/10.1101/2020.05.06.20092700
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA et al (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama 278(16):1349–1356
Jivanji CJ, Asrani VM, Windsor JA, Petrov MS (2017) New-onset diabetes after acute and critical illness: a systematic review. Mayo Clin Proc 92(5):762–773. https://doi.org/10.1016/j.mayocp.2016.12.020
Kuo CL, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA, Melzer D (2020) APOE e4 genotype predicts severe COVID-19 in the UK Biobank community cohort. J Gerontol A Biol Sci Med Sci. https://doi.org/10.1093/gerona/glaa131
Yang JK, Lin SS, Ji XJ, Guo LM (2010) Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol 47(3):193–199. https://doi.org/10.1007/s00592-009-0109-4
Kehoe PG, Wong S, Al Mulhim N, Palmer LE, Miners JS (2016) Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-β and tau pathology. Alzheimers Res Ther 8(1):50. https://doi.org/10.1186/s13195-016-0217-7
Jiang T, Zhang YD, Zhou JS, Zhu XC, Tian YY, Zhao HD, Lu H, Gao Q et al (2016) Angiotensin-(1-7) is reduced and inversely correlates with tau hyperphosphorylation in animal models of Alzheimer’s disease. Mol Neurobiol 53(4):2489–2497. https://doi.org/10.1007/s12035-015-9260-9
Fekih-Mrissa N, Bedoui I, Sayeh A, Derbali H, Mrad M, Mrissa R, Nsiri B (2017) Association between an angiotensin-converting enzyme gene polymorphism and Alzheimer’s disease in a Tunisian population. Ann General Psychiatry 16:41. https://doi.org/10.1186/s12991-017-0164-0
Yan Y-Q, Fang Y, Zheng R, Pu J-L, Zhang B-R (2020) NLRP3 Inflammasomes in Parkinson’s disease and their regulation by Parkin. Neuroscience 446:323–334. https://doi.org/10.1016/j.neuroscience.2020.08.004
Corrêa DG, de Souza Lima FC, da Cruz BD, Coutinho AC, Hygino da Cruz LC (2020) COVID-19 associated with encephalomyeloradiculitis and positive anti-aquaporin-4 antibodies: cause or coincidence? Mult Scler J. https://doi.org/10.1177/1352458520949988
Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong J-C, Turner AJ, Raizada MK, Grant MB et al (2020) Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system. Circ Res 126(10):1456–1474. https://doi.org/10.1161/CIRCRESAHA.120.317015
Inoue Y, Tanaka N, Tanaka Y, Inoue S, Morita K, Zhuang M, Hattori T, Sugamura K (2007) Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J Virol 81(16):8722–8729. https://doi.org/10.1128/jvi.00253-07
Mao XY, Jin WL (2020) The COVID-19 pandemic: consideration for brain infection. Neuroscience 437:130–131. https://doi.org/10.1016/j.neuroscience.2020.04.044
Beach SR, Praschan NC, Hogan C, Dotson S, Merideth F, Kontos N, Fricchione GL, Smith FA (2020) Delirium in COVID-19: a case series and exploration of potential mechanisms for central nervous system involvement. Gen Hosp Psychiatry 65:47–53. https://doi.org/10.1016/j.genhosppsych.2020.05.008
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–1418. https://doi.org/10.1016/S0140-6736(20)30937-5
Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, Ueno M, Sakata H et al (2020) A first case of meningitis/encephalitis associated with SARS-coronavirus-2. Int J Infect Dis 94:55–58. https://doi.org/10.1016/j.ijid.2020.03.062
Edén A, Kanberg N, Gostner J, Fuchs D, Hagberg L, Andersson L-M, Lindh M, Price RW, Zetterberg H, Gisslén M (2020) CSF biomarkers in patients with COVID-19 and neurological symptoms. A case series https://doi.org/10.1212/WNL.0000000000010977
Bostancıklıoğlu M (2020) SARS-CoV2 entry and spread in the lymphatic drainage system of the brain. Brain Behav Immun 87:122–123. https://doi.org/10.1016/j.bbi.2020.04.080
Bagheri SHR, Asghari AM, Farhadi M, Shamshiri AR, Kabir A, Kamrava SK, Jalessi M, Mohebbi A, Alizadeh R, Honarmand AA, Ghalehbaghi B, Salimi A (2020) Coincidence of COVID-19 epidemic and olfactory dysfunction outbreak. medRxiv:2020.2003.2023.20041889. doi:https://doi.org/10.1101/2020.03.23.20041889
Eliezer M, Hamel A-L, Houdart E, Herman P, Housset J, Jourdaine C, Eloit C, Verillaud B et al (2020) Loss of smell in COVID-19 patients: MRI data reveals a transient edema of the olfactory clefts. Neurology. https://doi.org/10.1212/wnl.0000000000010806
Chen R, Wang K, Yu J, Chen Z, Wen C, Xu Z (2020) The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain. bioRxiv:2020.2004.2007.030650. doi:https://doi.org/10.1101/2020.04.07.030650
Shiers S, Ray PR, Wangzhou A, Tatsui CE, Rhines L, Li Y, Uhelski ML, Dougherty PM, Price TJ (2020) ACE2 expression in human dorsal root ganglion sensory neurons: implications for SARS-CoV-2 virus-induced neurological effects. bioRxiv:2020.2005.2028.122374. doi:https://doi.org/10.1101/2020.05.28.122374
Costello F, Dalakas MC (2020) Cranial neuropathies and COVID-19: neurotropism and autoimmunity. Neurology. https://doi.org/10.1212/WNL.0000000000009921
Esposito G, Pesce M, Seguella L, Sanseverino W, Lu J, Sarnelli G (2020) Can the enteric nervous system be an alternative entrance door in SARS-CoV2 neuroinvasion? Brain Behav Immun 87:93–94. https://doi.org/10.1016/j.bbi.2020.04.060
Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJ, Kraneveld AD (2017) Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol 8:37. https://doi.org/10.3389/fneur.2017.00037
Paniz-Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J, Sordillo EM, Fowkes M (2020) Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol 92(7):699–702. https://doi.org/10.1002/jmv.25915
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S (2008) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol 82(15):7264–7275. https://doi.org/10.1128/jvi.00737-08
Ye M, Ren Y, Lv T (2020) Encephalitis as a clinical manifestation of COVID-19. Brain Behav Immun. https://doi.org/10.1016/j.bbi.2020.04.017
Duong L, Xu P, Liu A (2020) Meningoencephalitis without respiratory failure in a young female patient with COVID-19 infection in Downtown Los Angeles, early April 2020. Brain Behav Immun 87:33. https://doi.org/10.1016/j.bbi.2020.04.024
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506. https://doi.org/10.1016/s0140-6736(20)30183-5
Benameur K, Agarwal A, Auld S, Butters M, Webster A, Ozturk T, Howell JC, Bassit L et al (2020) Encephalopathy and encephalitis associated with cerebrospinal fluid cytokine alterations and coronavirus disease, Atlanta, Georgia, USA, 2020. Emerg Infect Dis J 26(9):2016–2021. https://doi.org/10.3201/eid2609.202122
Yarlagadda A, Alfson E, Clayton AH (2009) The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry (Edgmont) 6(11):18–22
Dufek M, Rektorova I, Thon V, Lokaj J, Rektor I (2015) Interleukin-6 may contribute to mortality in Parkinson’s disease patients: a 4-year prospective study. Parkinson Disease 2015:898192. https://doi.org/10.1155/2015/898192
Lindlau A, Widmann CN, Putensen C, Jessen F, Semmler A, Heneka MT (2015) Predictors of hippocampal atrophy in critically ill patients. Eur J Neurol 22(2):410–415. https://doi.org/10.1111/ene.12443
Heneka MT, Golenbock D, Latz E, Morgan D, Brown R (2020) Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res Ther 12(1):69. https://doi.org/10.1186/s13195-020-00640-3
Domingues C, da Cruz ESOAB, Henriques AG (2017) Impact of cytokines and chemokines on Alzheimer’s disease neuropathological hallmarks. Curr Alzheimer Res 14(8):870–882. https://doi.org/10.2174/1567205014666170317113606
Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, Propson NE, Xu Y et al (2020) Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest 130(4):1912–1930. https://doi.org/10.1172/jci133737
Jones HD, Crother TR, Gonzalez-Villalobos RA, Jupelli M, Chen S, Dagvadorj J, Arditi M, Shimada K (2014) The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol 50(2):270–280. https://doi.org/10.1165/rcmb.2013-0087OC
Palao M, Fernández-Díaz E, Gracia-Gil J, Romero-Sánchez CM, Díaz-Maroto I, Segura T (2020) Multiple sclerosis following SARS-CoV-2 infection. Mult Scler Relat Disord 45:102377. https://doi.org/10.1016/j.msard.2020.102377
Fazzini E, Fleming J, Fahn S (1992) Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson’s disease. Mov Disord 7(2):153–158. https://doi.org/10.1002/mds.870070210
Desforges M, Le Coupanec A, Dubeau P, Bourgouin A, Lajoie L, Dubé M, Talbot PJ (2019) Human coronaviruses and other respiratory viruses: underestimated opportunistic pathogens of the central nervous system? Viruses 12(1). https://doi.org/10.3390/v12010014
Merad M, Martin JC (2020) Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 20(6):355–362. https://doi.org/10.1038/s41577-020-0331-4
Sevenich L (2018) Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol 9:697. https://doi.org/10.3389/fimmu.2018.00697
Desforges M, Miletti TC, Gagnon M, Talbot PJ (2007) Activation of human monocytes after infection by human coronavirus 229E. Virus Res 130(1-2):228–240. https://doi.org/10.1016/j.virusres.2007.06.016
Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, Chng MHY, Lin M et al (2020) SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 584(7821):457–462. https://doi.org/10.1038/s41586-020-2550-z
Swadling L, Maini MK (2020) T cells in COVID-19—united in diversity. Nat Immunol. https://doi.org/10.1038/s41590-020-0798-y
Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D et al (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119(1):182–192. https://doi.org/10.1172/jci36470
Zheng Y, Liu X, Le W, Xie L, Li H, Wen W, Wang S, Ma S et al (2020) A human circulating immune cell landscape in aging and COVID-19. Protein Cell 11(10):740–770. https://doi.org/10.1007/s13238-020-00762-2
Dolatshahi M, Pourmirbabaei S, Kamalian A, Ashraf-Ganjouei A, Yaseri M, Aarabi MH (2018) Longitudinal alterations of alpha-synuclein, amyloid beta, total, and phosphorylated tau in cerebrospinal fluid and correlations between their changes in Parkinson’s disease. Front Neurol 9:560
Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, Kehrl JH (2014) SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol 193(6):3080–3089. https://doi.org/10.4049/jimmunol.1303196
Ye Z, Wong CK, Li P, Xie Y (2008) A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim Biophys Acta 1780(12):1383–1387. https://doi.org/10.1016/j.bbagen.2008.07.009
Aoe T (2020) Pathological aspects of COVID-19 as a conformational disease and the use of pharmacological chaperones as a potential therapeutic strategy. Front Pharmacol 11:1095. https://doi.org/10.3389/fphar.2020.01095
Sureda A, Alizadeh J, Nabavi SF, Berindan-Neagoe I, Cismaru CA, Jeandet P, Łos MJ, Clementi E et al (2020) Endoplasmic reticulum as a potential therapeutic target for COVID-19 infection management? Eur J Pharmacol 882:173288. https://doi.org/10.1016/j.ejphar.2020.173288
Wang Y, Xu E, Musich PR, Lin F (2019) Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther 25(7):816–824. https://doi.org/10.1111/cns.13116
Zhang H, Liao YS, Gong J, Liu J, Xia X, Zhang H (2020) Clinical characteristics of coronavirus disease (COVID-19) patients with gastrointestinal symptoms: a report of 164 cases. Dig Liver Dis. https://doi.org/10.1016/j.dld.2020.04.034
Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H, Wan Y, Chung A et al (2020) Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology. https://doi.org/10.1053/j.gastro.2020.05.048
Ambrosini YM, Borcherding D, Kanthasamy A, Kim HJ, Willette AA, Jergens A, Allenspach K, Mochel JP (2019) The gut-brain axis in neurodegenerative diseases and relevance of the canine model: a review. Front Aging Neurosci 11:130. https://doi.org/10.3389/fnagi.2019.00130
Dhar D, Mohanty A (2020) Gut microbiota and Covid-19-possible link and implications. Virus Res 285:198018. https://doi.org/10.1016/j.virusres.2020.198018
Abiodun OA, Ola MS (2020) Role of brain renin angiotensin system in neurodegeneration: An update. Saudi J Biol Sci 27(3):905–912. https://doi.org/10.1016/j.sjbs.2020.01.026
Nataf S (2020) An alteration of the dopamine synthetic pathway is possibly involved in the pathophysiology of COVID-19. J Med Virol. https://doi.org/10.1002/jmv.25826
Benetti E, Tita R, Spiga O, Ciolfi A, Birolo G, Bruselles A, Doddato G, Giliberti A et al (2020) ACE2 gene variants may underlie interindividual variability and susceptibility to COVID-19 in the Italian population. Eur J Hum Genet. https://doi.org/10.1038/s41431-020-0691-z
Devaux CA, Rolain J-M, Raoult D (2020) ACE2 receptor polymorphism: Susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J Microbiol Immunol Infect 53(3):425–435. https://doi.org/10.1016/j.jmii.2020.04.015
Lin JJ, Yueh KC, Chang DC, Lin SZ (2002) Association between genetic polymorphism of angiotensin-converting enzyme gene and Parkinson’s disease. J Neurol Sci 199(1-2):25–29. https://doi.org/10.1016/s0022-510x(02)00081-3
Kaur P, Muthuraman A, Kaur M (2015) The implications of angiotensin-converting enzymes and their modulators in neurodegenerative disorders: current and future perspectives. ACS Chem Neurosci 6(4):508–521. https://doi.org/10.1021/cn500363g
Sierra-Fonseca JA, Gosselink KL (2018) Tauopathy and neurodegeneration: a role for stress. Neurobiol Stress 9:105–112. https://doi.org/10.1016/j.ynstr.2018.08.009
Ibrahimagic OC, Jakubovic AC, Smajlovic D, Dostovic Z, Kunic S, Iljazovic A (2016) Psychological stress and changes of hypothalamic-pituitary-adrenal axis in patients with “de novo” Parkinson’s disease. Mediev Archaeol 70(6):445–448. https://doi.org/10.5455/medarh.2016.70.445-448
Agarwal S, Agarwal SK (2020) Endocrine changes in SARS-CoV-2 patients and lessons from SARS-CoV. Postgrad Med J 96(1137):412–416. https://doi.org/10.1136/postgradmedj-2020-137934
Oudit GY, Kassiri Z, Jiang C, Liu PP, Poutanen SM, Penninger JM, Butany J (2009) SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Investig 39(7):618–625. https://doi.org/10.1111/j.1365-2362.2009.02153.x
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P et al (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat Med 11(8):875–879. https://doi.org/10.1038/nm1267
Wang LA, de Kloet AD, Smeltzer MD, Cahill KM, Hiller H, Bruce EB, Pioquinto DJ, Ludin JA et al (2018) Coupling corticotropin-releasing-hormone and angiotensin converting enzyme 2 dampens stress responsiveness in male mice. Neuropharmacology 133:85–93. https://doi.org/10.1016/j.neuropharm.2018.01.025
Steenblock C, Todorov V, Kanczkowski W, Eisenhofer G, Schedl A, Wong M-L, Licinio J, Bauer M et al (2020) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the neuroendocrine stress axis. Mol Psychiatry. https://doi.org/10.1038/s41380-020-0758-9
Leow MK, Kwek DS, Ng AW, Ong KC, Kaw GJ, Lee LS (2005) Hypocortisolism in survivors of severe acute respiratory syndrome (SARS). Clin Endocrinol 63(2):197–202. https://doi.org/10.1111/j.1365-2265.2005.02325.x
Morris G, Anderson G, Maes M (2017) Hypothalamic-pituitary-adrenal hypofunction in myalgic encephalomyelitis (ME)/chronic fatigue syndrome (CFS) as a consequence of activated immune-inflammatory and oxidative and nitrosative pathways. Mol Neurobiol 54(9):6806–6819. https://doi.org/10.1007/s12035-016-0170-2
Chen W (2020) A potential treatment of COVID-19 with TGF-β blockade. Int J Biol Sci 16(11):1954–1955. https://doi.org/10.7150/ijbs.46891
Rodriguez-Morales AJ, Cardona-Ospina JA, Gutiérrez-Ocampo E, Villamizar-Peña R, Holguin-Rivera Y, Escalera-Antezana JP, Alvarado-Arnez LE, Bonilla-Aldana DK et al (2020) Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med Infect Dis 34:101623. https://doi.org/10.1016/j.tmaid.2020.101623
Sasannejad C, Ely EW, Lahiri S (2019) Long-term cognitive impairment after acute respiratory distress syndrome: a review of clinical impact and pathophysiological mechanisms. Crit Care 23(1):352. https://doi.org/10.1186/s13054-019-2626-z
Kanberg N, Ashton NJ, Andersson LM, Yilmaz A, Lindh M, Nilsson S, Price RW, Blennow K et al (2020) Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology. https://doi.org/10.1212/wnl.0000000000010111
Fries M, Bickenbach J, Henzler D, Beckers S, Dembinski R, Sellhaus B, Rossaint R, Kuhlen R (2005) S-100 protein and neurohistopathologic changes in a porcine model of acute lung injury. Anesthesiology 102(4):761–767. https://doi.org/10.1097/00000542-200504000-00011
Erickson MA, Banks WA (2013) Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab 33(10):1500–1513. https://doi.org/10.1038/jcbfm.2013.135
Sonneville R, Verdonk F, Rauturier C, Klein IF, Wolff M, Annane D, Chretien F, Sharshar T (2013) Understanding brain dysfunction in sepsis. Ann Intensive Care 3(1):15. https://doi.org/10.1186/2110-5820-3-15
Taccone FS, Castanares-Zapatero D, Peres-Bota D, Vincent JL, Berre J, Melot C (2010) Cerebral autoregulation is influenced by carbon dioxide levels in patients with septic shock. Neurocrit Care 12(1):35–42. https://doi.org/10.1007/s12028-009-9289-6
Hess DC, Eldahshan W, Rutkowski E (2020) COVID-19-related stroke. Transl Stroke Res 11(3):322–325. https://doi.org/10.1007/s12975-020-00818-9
Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J et al (2020) Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb Res 191:148–150. https://doi.org/10.1016/j.thromres.2020.04.041
Xu P, Sriramula S, Lazartigues E (2011) ACE2/ANG-(1–7)/Mas pathway in the brain: the axis of good. Am J Phys Regul Integr Comp Phys 300(4):R804–R817. https://doi.org/10.1152/ajpregu.00222.2010
Scudamore O, Ciossek T (2018) Increased oxidative stress exacerbates α-synuclein aggregation in vivo. J Neuropathol Exp Neurol 77(6):443–453. https://doi.org/10.1093/jnen/nly024
Goldstein MR, Poland GA, Graeber WC (2020) Does apolipoprotein E genotype predict COVID-19 severity? QJM: an International Journal of Medicine. https://doi.org/10.1093/qjmed/hcaa142
Wang H, Yuan Z, Pavel MA, Hansen SB (2020) The role of high cholesterol in age-related COVID19 lethality. bioRxiv. https://doi.org/10.1101/2020.05.09.086249
Riedel BC, Thompson PM, Brinton RD (2016) Age, APOE and sex: triad of risk of Alzheimer’s disease. J Steroid Biochem Mol Biol 160:134–147. https://doi.org/10.1016/j.jsbmb.2016.03.012
Wu Q, Zhou L, Sun X, Yan Z, Hu C, Wu J, Xu L, Li X et al (2017) Altered lipid metabolism in recovered SARS patients twelve years after infection. Sci Rep 7(1):9110. https://doi.org/10.1038/s41598-017-09536-z
Huang M, Parker AM, Bienvenu OJ, Dinglas VD, Colantuoni E, Hopkins RO, Needham DM (2016) psychiatric symptoms in acute respiratory distress syndrome survivors: a 1-year national multicenter study. Crit Care Med 44(5):954–965. https://doi.org/10.1097/ccm.0000000000001621
Higgs S, Spetter MS (2018) Cognitive Control of eating: the role of memory in appetite and weight gain. Curr Obes Rep 7(1):50–59. https://doi.org/10.1007/s13679-018-0296-9
Cai H, Cong WN, Ji S, Rothman S, Maudsley S, Martin B (2012) Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr Alzheimer Res 9(1):5–17. https://doi.org/10.2174/156720512799015064
Kang Z, Luo S, Gui Y, Zhou H, Zhang Z, Tian C, Zhou Q, Wang Q et al (2020) Obesity is a potential risk factor contributing to clinical manifestations of COVID-19. Int J Obes. https://doi.org/10.1038/s41366-020-00677-2
Cao X, Yin R, Albrecht H, Fan D, Tan W (2020) Cholesterol: a new game player accelerating vasculopathy caused by SARS-CoV-2? Am J Physiol Endocrinol Metab 319(1):E197–e202. https://doi.org/10.1152/ajpendo.00255.2020
Hribar CA, Cobbold PH, Church FC (2020) Potential role of vitamin D in the elderly to resist COVID-19 and to slow progression of Parkinson’s disease. Brain Sci 10(5):284. https://doi.org/10.3390/brainsci10050284
Author information
Authors and Affiliations
Contributions
M.D. and MH.A contributed to forming the idea and designing the outline of the article; M.D. and MM.S. contributed to writing the first draft and revising it; MM.S. and M.D. designed the graphical abstract.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
Not applicable
Consent to Participate
Not applicable
Consent for Publication
All authors declare their consent for publication
Code Availability
Not applicable
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: There is an error on affiliation of Dr. MohammadHadi Aarabi.
Rights and permissions
About this article

Cite this article
Dolatshahi, M., Sabahi, M. & Aarabi, M.H. Pathophysiological Clues to How the Emergent SARS-CoV-2 Can Potentially Increase the Susceptibility to Neurodegeneration. Mol Neurobiol 58, 2379–2394 (2021). https://doi.org/10.1007/s12035-020-02236-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02236-2