
Abstract
Similar to its predecessors, coronavirus disease 2019 (COVID-19) exhibits neurotrophic properties, which lead to progression of neurologic sequelae. Besides direct viral invasion to the central nervous system (CNS), indirect CNS involvement through viral-mediated immune response is plausible. Aberrant immune pathways such as extreme release of cytokines (cytokine storm), autoimmunity mediated by cross-reactivity between CNS components and viral particles, and microglial activation propagate CNS damage in these patients. Here, we review the currently available evidence to discuss the plausible immunologic pathways that may contribute to the development of COVID-19 neurological complications, namely Alzheimer’s disease, Parkinson’s disease, stroke, multiple sclerosis, Guillain-Barre syndrome, seizure, and brainstem involvement.
Similar content being viewed by others

Introduction
In December 2019, the outbreak of novel coronavirus disease 2019 (COVID-19) emerged in Wuhan, China. To date, the outbreak has turned into a pandemic, infecting millions of people globally [1]. Although the virus, also known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), mainly manifests as an acute respiratory infection [2], recent evidence suggests that 36% of affected patients exhibit neurological sequelae [3]. Neurologic symptoms are more common among severe cases of the disease, regarding that 84% of the intensive care unit (ICU)–admitted SARS-CoV-2 patients exhibited at least one neurologic symptom [4]. In line with these, brain magnetic resonance imaging (MRI) investigations in SARS-CoV-2 patients show multifocal hyperintense white matter lesions and cortical signal abnormalities (particularly in the medial temporal lobe) on fluid-attenuated inversion recovery (FLAIR), along with intracerebral hemorrhagic and microhemorrhagic lesions, and leptomeningeal enhancement [5, 6]. Electroencephalogram (EEG) studies demonstrate high amplitude monomorphic delta waves, indicating the central nervous system (CNS) involvement in SARS-CoV-2 patients [7]. Also, functional magnetic resonance imaging (fMRI) is recently proposed as an independent predictor of neurologic outcomes [8]. SARS-CoV-2 patients present with elevated plasma levels of neurofilament light chain protein (NfL) and glial fibrillary acidic protein (GFAP), which are known as biochemical indicators of neuronal injury and glial activation, respectively [9]. Also, postmortem brain autopsies demonstrate virus invasion to different brain regions, including the hypothalamus and olfactory bulb, accompanied by neural death and demyelination [10, 11].
Several pathogenic mechanisms have been proposed for the neurological deficit in SARS-CoV-2: indirect CNS involvement through systemic inflammation, direct invasion of the virus into the CNS, multi-organ failure, hypoxia, sepsis, etc. [12, 13]. In this article, we aim to revisit the possible pathomechanisms that may contribute to development of neurologic complications in patients afflicted with this virus with a substantial focus on immunological pathways.
Pathomechanisms of Immune-Mediated CNS Involvement by COVID
Regarding the fact that SARS-CoV-2 exhibits a 75–80% genome sequence similarity to its predecessors, namely severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) [13], we first discuss the immunologic aspects of neurologic sequelae for the SARS-CoV and MERS-CoV.
SARS-CoV
Severe acute respiratory syndrome (SARS) caused by SARS-CoV emerged in Asia in 2003 and mainly manifested as an acute respiratory infection. However, studies demonstrate that SARS-CoV associates with neurological deficits like stroke, encephalopathy, seizure, Guillain-Barre syndrome (GBS), and brainstem involvement [14,15,16].
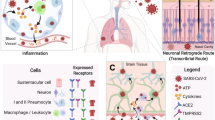
Several findings reinforce the idea of CNS involvement in SARS-CoV: first, postmortem SARS-CoV patients mostly show cerebral edema and meningeal vasodilation in autopsy investigations [17]. Second, SARS-CoV genome sequences are found in cerebrospinal fluid (CSF) and throughout the hypothalamus and cortex of infected patients [17, 18]. Third, in vitro studies demonstrate that human glial cells, as well as brain endothelial and smooth muscle cells, express angiotensin-converting enzyme 2 (ACE-2) [19]. Further investigations in rodents show that ACE-2 is expressed in brain regions like cortex and raphe and brainstem, predominantly in neurons [19]. Also, spatial distribution analysis reveals that ACE-2 is highly expressed by neurons and glial cells in different human brain regions, including middle temporal gyrus, cingulate gyrus, substantia nigra, choroid plexus, and, to a lesser extent, hippocampus [20]. ACE-2 is a type I transmembrane metallocarboxypeptidase that acts as a receptor for SARS-CoV as well as SARS-CoV-2 [21]. Binding of viral spike (S) protein to ACE-2 receptors accompanied by proteolytic cleavage of viral spike protein mediated by transmembrane serine protease 2 (TMPRSS2) facilitates cell entry (Fig. 1) [21].

Three phases of CNS infection by SARS-CoV-2. First, the virus directly invades the brain through vascular or retrograde axonal transport within the cribriform plate, and thus the viral load increases in the CSF. Second, the virus infects, replicates in, and kills neural cells through ACE-2 receptors and TMPRSS2, a facilitator of viral entry. Finally, in the third phase, the immune-mediated response against the virus can indirectly involve the brain, although the viral replication declines in this last phase. CNS central nervous system, ACE-2 angiotensin-converting enzyme 2, TMPRSS2 transmembrane serine protease 2. Adopted from https://www.dpz.eu/de/infothek/wissen/coronaviren.html, by Markus Hoffmann. Adopted with permission
Systemic inflammatory response syndrome (SIRS) is defined as excessive host immune response against noxious stimuli (e.g., viral infection), through which the primary protective role of cytokine release turns into a detrimental response against host tissues, leading to impaired integrity of capillary walls and end-organ dysfunction [22]. SIRS accounts for the neurologic sequelae found in SARS-CoV patients [23]. SARS-CoV infects macrophages, monocytes, and dendritic cells and upregulates the expression of pro-inflammatory cytokines (e.g., tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6)), and inflammatory chemokines (C-C motif chemokine ligand 2 (CCL2), C-C motif chemokine ligand 3 (CCL3), C-C motif chemokine ligand 5 (CCL5), C-X-C motif chemokine 10 (CXCL10)) [24]. Complicated cases of SARS-CoV exhibit higher levels of pro-inflammatory cytokines like IL-6 and interferon-γ (IFN-γ), making them more susceptible to neurologic complications [25]. Studies on postmortem cases indicate that lymphocytes and monocytes infiltrate in brain vessel walls, exacerbating the neuronal degeneration and demyelination process [17]. Last but not least, indirect immunofluorescence staining and cell-based enzyme-linked immunosorbent assay (ELISA) study in the serum of SARS-CoV patients reveal the presence of IgG and IgM autoantibodies as well as complement cytotoxicity against human epithelial and endothelial cells [26]. This finding implies a role for autoimmunity in post-infectious complications of SARS-CoV.
MERS-CoV
Middle East respiratory syndrome (MERS), another respiratory infection from the CoV family, was first reported in Saudi Arabia in 2012. Nearly 20% of MERS-CoV patients develop neurologic manifestations during the disease course [17]. However, the neurologic sequelae do not always occur in the infection process and might manifest after a delay of 2 or 3 weeks [17]. Stroke, encephalopathy, seizure, GBS, and brainstem involvement could accompany MERS infection [16, 17]. Brain MRI in neurologically affected MERS-CoV patients reveal hyperintense lesions in white matter and subcortical frontal, parietal, and temporal lobes, along with basal ganglia and corpus callosum [27]. The mechanisms of neurological involvement in MERS-CoV are almost similar to SARS-CoV, with a surge in circulating cytokine levels and a role for autoreactive autoantibodies [28, 29].
SARS-CoV-2
SARS-CoV-2 invasion throughout the CNS can be divided into three phases: first, the virus directly invades the brain through vascular or retrograde axonal transport within the cribriform plate, and thus, the viral load increases in the CSF [4]. Second, the virus infects, replicates in, and kills neural cells through ACE-2 receptors [30]. Third, the immune-mediated response against the virus can indirectly involve the brain, although the viral replication declines in this final phase [31] (Fig. 1).
Recent evidence shows that the third phase (also known as SIRS) accounts for the majority of CNS disturbances mediated by SARS-CoV-2 [31]. SARS-CoV-2 invades astrocytes, macrophages, and microglia in CNS and induces a pro-inflammatory state characterized by increased levels of inflammatory mediators including interleukin-1β (IL-1 β), interleukin-2 (IL-2), IL-2 receptor (IL-2R), interleukin-4 (IL-4), IL-6, interleukin-10 (IL-10), interleukin-18 (IL-18), IFN-γ, TNF-α, granulocyte colony-stimulating factor (GCSF), monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 1-α (MIP-1α), CXCL10, CCL2, and C-reaction protein (CRP) [25]. Also, SARS-CoV-2 induces in vitro expression of inflammatory cytokines such as IL-6, interleukin-12 (IL-12), interleukin-15 (IL-15), and TNF-α in glial cells [17]. Severe cases of the disease compared with non-severe cases tend to increase in circulatory levels of IL-2, IL-2R, IL-6, IL-10, IFN-γ, TNF-α, CCL2, procalcitonin (PCT), CRP, erythrocyte sedimentation rate (ESR), and white blood cell (WBC) and neutrophil counts and decrease in total lymphocyte, CD4+ T lymphocyte, and CD8+ T lymphocyte counts, while B cell count remains in the normal range [24, 32, 33]. Also, antibodies (namely IgG, IgM, and IgA) against viral particles rise in the disease course [34]. Seroconversion of IgM and IgG antibodies begins 4 days after the symptom onset [35]. Although the median time for seropositivity against the receptor-binding domain of the virus in the serum is equal for different isotypes, IgG is estimated to remain positive in the serum for 75 days, that is the longest period comparing with IgA and IgM, lasting for 51 and 47 days, respectively [34]. Of note, the social isolation and the resultant stress exacerbate the aforementioned cytokine release in affected patients [25].
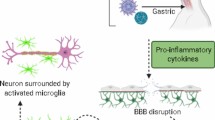
Cytokine storm is a term used for the extreme release of interferons, tumor necrosis factors, chemokines, interleukins, and other mediators in a hyperactive and injurious manner against host tissues [36]. Cytokine storm is attributed to SARS-CoV-2 as well as SARS-CoV infection and accompany with a spectrum of SIRS manifestations, including hypotension, tachycardia, tachypnea, and fever, leading to adverse clinical outcomes [36]. Cytokine storm involves the CNS, regarding the fact that most cytokines are either produced in the brain tissue or able to cross the blood-brain barrier in the condition that released cytokines (namely, IL-1β, IL-6, and TNF-α) disrupt the integrity of blood-brain barrier [37]. So, brain autopsy studies of postmortem SARS-CoV-2 patients demonstrate the infiltration of monocytes, macrophages, and T-lymphocytes into the vessel walls (Fig. 2) [15]. Also, interferon-α2 (IFN-α2) and IFN-γ upregulate the expression of ACE-2 receptors and, thus, facilitate the entry of the virus into host cells in-vitro [38]. Cytokine storm results in neural death, activation of microglia, disruption of synaptic plasticity, and impairment in neurotransmitter metabolism. Cytokine storm is also a predictor of hippocampal atrophy [12]. Cytokine storm stimulates the secretion of glucocorticoids through manipulation of the hypothalamic-pituitary-adrenocortical axis; nevertheless, the resultant increase in glucocorticoids is not able to suppress the inflammation due to impaired negative feedback mechanism [25]. Last but not least, the cytokine storm might lead to delayed immune dysregulation after the disease course, manifested by persistent inflammation or immunosuppression, which might cause delayed complications [25]. CNS features of SARS-CoV, MERS-CoV, and SARS-CoV-2 are summarized in Table 1.

WBC infiltration and subsequent CNS damage mediated by SARS-CoV-2. Cytokine storm involves the CNS, and the released cytokines disrupt the integrity of blood-brain barrier. As a result, monocytes and lymphocytes infiltrate into the vessel walls and lead to neuroinflammation, neurodegeneration, and demyelination. CNS central nervous system, WBC white blood cell
Regarding the widespread influence of SARS-CoV-2-mediated immune response on CNS, we discuss the inflammatory correlates of neurologic sequelae of SARS-CoV-2 in the following sections.
Pathomechanisms of Immune-Mediated CNS Complications of SARS-CoV-2
Brain areas like the hippocampus and midbrain are vulnerable to direct SARS-CoV-2 invasion and induction of Alzheimer’s disease (AD) and Parkinson’s disease (PD), respectively [18]. The aberrant immune response characterized by a surge in cytokine levels (e.g., IL-6) derived by SARS-CoV-2 accelerates the process of neurodegeneration that may contribute to the development of neurodegenerative diseases [42]. The mentioned aberrant immune response may be a consequence of infection of intestinal mucosa and modulation of the gut microbiome by SARS-CoV-2, which can trigger neuroinflammation and neurodegeneration [24]. Interestingly, modulation of the gut microbiome plays a role in the pathophysiology of AD and PD [43, 44]. Seemingly, other coronaviruses like human coronavirus OC43 (HCV-OC43) may trigger delayed neural degeneration in animal models [45].
Alzheimer’s Disease
AD, the most prevalent neurodegenerative disorder, is characterized by the accumulation of intraneuronal aggregates of hyperphosphorylated tau and extracellular beta-amyloid plaques [37, 46, 47]. Considering the role of cholesterol-binding protein (Apolipoprotein E (ApoE)), a potential link between AD and SARS-CoV-2 infection is implicated. Serum cholesterols bind to ApoE receptors and induce ACE-2 receptor transport to the cell surface [48]. Super-resolution imaging study demonstrates that high cholesterol levels confer a 2-fold increase in SARS-CoV-2 entry sites and thus facilitate the viral entry [48]. ApoE and ACE-2 are highly expressed in alveolar type II cells, and the ApoE e4 allele modulates the pro-/anti-inflammatory state in macrophages [49]. On the other hand, ApoE, acting in conjunction with APOC1 and APOJ, transports cholesterol to maintain the myelin and neural membranes, dendritic reorganization, and synaptic turnover [50]. ApoE functions as a beta-amyloid chaperone through the transport of beta-amyloids to the lysosomes [50]. ApoE e4 allele associates with increased formation and deposition of beta-amyloid, and ApoE e4-positive mice exhibit increased accumulation of tau aggregates [50]. In line with these, homozygote ApoE e4e4 not only acts as a risk factor for severe SARS-CoV-2 infection but also confers a 14-fold increase in susceptibility to AD [49].
A growing body of evidence implicates that neuroinflammation may be an origin of AD. Persistent systemic inflammation can activate glial cells like microglia and astrocytes. Activated microglia secrete pro-inflammatory cytokines (e.g., IL-1β, IL-6, IL-12, TNF-α) [37, 51]. We hypothesize that not only the persistent systemic inflammation caused by SARS-CoV-2 may act as a trigger for microglial activation but also large amounts of pro-inflammatory cytokines secreted in response to this viral infection may aggravate neurodegeneration leading to AD. Elevated levels of TNF-α associate with a 4-fold increase in cognitive dysfunction [37]. Increased IL-1β and IL-6 mitigates microglial phagocytosis of β-amyloid plaques and thus impairs synaptic plasticity and memory [19, 25]. IFN induces synaptic degeneration as a mediator of post-viral inflammatory response. It is worth knowing that β-amyloids can encircle viral particles and enhance the aforementioned IFN-mediated response [52]. On the other hand, NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasomes play a role in microglia-mediated IL-1β release in AD. SARS-CoV-2 open reading frame 3a (ORF3a) protein stimulates NLRP3 inflammasomes, activation of which accelerates the neurodegeneration process in AD [12].
Parkinson’s Disease
PD, the second most common neurodegenerative disorder, is characterized by the degeneration of dopaminergic neurons of substantia nigra pars compacta, along with microscopic findings of intracellular α-synuclein aggregates [53,54,55,56]. Autoantibodies against α-synuclein, gangliosides, pigment neuromelanin Ma1/Ma2, leucine-rich glioma inactivated 1 (LGI1), and glutamic acid decarboxylase (GAD65) are found in the serum of PD patients, which implicate a role for autoimmunity in the pathogenesis of PD [53]. Knowing that SARS-CoV-2 may remain latent in neurons, we hypothesize that autoimmunity in SARS-CoV-2 infection due to cross-reactivity mediated by antiviral antibodies may lead to delayed progression of PD [28]. This hypothesis is supported by the fact that anti-CoV antibodies are found in the CSF of PD patients [28]. We also postulate that aberrant cytokine release into the CSF of SARS-CoV-2 patients may increase the risk of progression into PD by aggravating the neurodegeneration process, considering the fact that PD patients exhibit elevated levels of pro-inflammatory cytokines (including IL-1β, IL-6, TNF-α, and IFN-γ) in CSF. Cytokine storm activates microglia, which are implicated in the pathophysiology of PD [57]. Moreover, the extreme release of cytokines into the CSF mediated by SARS-CoV-2 dysregulates the balance between production, release, and reuptake of neurotransmitters including dopamine, which possibly contributes to the development of PD [25].
Moreover, the common finding of anosmia (total loss of sense of smell) in PD and SARS-CoV-2 reinforces their association. Anosmia is a common finding in neurodegenerative diseases [58], and over 90% of PD patients develop anosmia or hyposmia (partial loss of sense of smell). Anosmia may precede motor symptoms of PD and thus can be used as a marker of underlying neurodegeneration [59]. Although MRI studies of PD patients show that volume and sulcus depth of olfactory bulb decrease, biopsy samples of olfactory epithelium are normal in these patents [60]. These findings suggest that the observed anosmia in these patients is due to a disturbance in central and not peripheral processing of smell [60]. Knowing that autopsy studies of postmortem PD patients suffering from anosmia show abundant deposition of Lewy bodies in the olfactory bulb, anosmia is attributed to the distribution of Lewy bodies from the medulla in early stages of PD [61]. However, in later stages, cognitive deficits and cholinergic denervation may underlie the development of anosmia in these patients [60].
On the other hand, a recent review reports a nearly 23% prevalence for anosmia in SARS-CoV-2 patients [62]. Several explanations are suggested to shed light on the underlying cause of anosmia in SARS-CoV-2 patients. At first, it was hypothesized that the local inflammation and the resulting coryza and sinusitis account for anosmia in these patients. However, recent imaging studies demonstrate no signs of sinusitis in SARS-CoV-2 patients. Moreover, the presence of anosmia in the absence of other symptoms suggests that coryza cannot be the reason for olfactory dysfunction in SARS-CoV-2 [15]. Indeed, milder inflammation and inflammatory infiltrations in SARS-CoV-2 infection comparing with SARS suggest a different route of infection through which the virus transmits via infected macrophages to the supporting cells of the olfactory epithelium and olfactory bulb, leading to impairment of olfactory receptors [63].
Several findings support this latter hypothesis: first, olfactory epithelial cells (particularly sustentacular cells) and olfactory bulb cells express ACE-2 and TMPRSS2 [64, 65]. Second, postmortem studies show that the virus can invade the brain through the olfactory epithelium and olfactory bulb [66]. Third, positron emission tomography–computed tomography (PET-CT) study in SARS-CoV-2 patients with anosmia demonstrate declined orbitofrontal cortex activity, suggesting neural dysfunction [67]. Forth, the genome sequence of the virus was found in the olfactory mucosa and olfactory bulb, indicating axonal transport [66]. The latter hypothesis, also known as post-viral olfactory dysfunction, is reported in a case of SARS-CoV as well and is more prevalent among patients with a history of the neurologic disease [64]. Maintenance of viral load in the olfactory epithelium in post-viral olfactory dysfunction can disrupt the regenerative capacity of neurons [64].
Stroke
Both ischemic and hemorrhagic strokes are being increasingly reported in SARS-CoV-2 patients and are the most common findings in neuroimaging studies (Table 2) [40, 75]. The prevalence of stroke is estimated to be between 0.2 and 1% of all infected patients [76], often proposed as the most common neurologic manifestation in SARS-CoV-2 patients [77]. High incidence of stroke in SARS-CoV-2 patients under 50 years old without cardiovascular risk factors accompanied by worse outcomes in SARS-CoV-2-mediated stroke comparing with other strokes indicates that this viral infection precipitates the pathways leading to stroke [76].
Postmortem autopsy studies of SARS-CoV-2-mediated stroke demonstrate hyperemic and edematous brain areas [76]. Also, micro- and macrothrombosis are found in other organs such as lung and kidney [76]. These findings indicate that coagulopathy may underlie the occurrence of stroke in these patients. Severe inflammatory response mediated by SARS-CoV-2 can upregulate pro-coagulative factors [17]. Recent investigations suggest that blood D-dimer, CRP, fibrinogen, and WBC count increases in SARS-CoV-2 patients, which propagate the hypercoagulable state. Moreover, a group of SARS-CoV-2 patients present positive lupus anticoagulant, anti-β2-glycoprotein-1, and anticardiolipin (marker of antiphospholipid syndrome) antibodies, which may precipitate stroke [15].
Another key contributor to stroke is the vascular wall injury, which induces the release of tissue factor [17]. Damage to vascular walls may occur as a consequence of direct viral invasion and replication in the vascular epithelium, also known as endotheliitis [66]. Endotheliitis due to SARS-CoV-2 is reported in other organs such as lung, heart, kidney, and bowel but not yet investigated in CNS [15]. Also, the aberrant immune response mediated by SARS-CoV-2 may provide the driving force for vascular wall damage [23]. This process takes place as a component of secondary hemophagocytic lymphohistiocytosis, characterized by inflammatory-mediated tissue damage through hyperactivation of macrophages and lymphocytes, leading to microangiopathy [15].
Several other mechanisms may increase the risk of stroke in SARS-CoV-2 infection. The cytokine storm mediated by SARS-CoV-2 precipitates microthrombosis [41]. In fact, elevated expression of pro-inflammatory cytokines (namely IL-1β, IL-6, and TNF-α) increases the risk of ischemic and hemorrhagic strokes [78]. Also, anti-cytokine therapies against these mediators protect from recurrence of stroke in experimental models [78]. Moreover, systemic inflammation can ignite atrial fibrillation and rupture the carotid plaques, leading to ischemic stroke [15]. Also, SARS-CoV-2 downregulates the expression of ACE-2 receptors, which degrade angiotensin II. So, the increase in angiotensin II and the resulting activation of the renin-angiotensin-aldosterone system (RAAS) system promotes sympathetic tone, oxidative stress, and inflammation and triggers fibrotic events [79].
Multiple Sclerosis
Multiple sclerosis (MS) is a chronic disabling disorder characterized by demyelination of white matter and, to a lesser extent, gray matter of CNS [51]. Recently, the first case-report of MS after SARS-CoV-2 infection was presented in the literature [80]. A SARS-CoV-2 patient without a history of neurological symptoms developed visual acuity and field defects and hyperreflexia. Orbital and brain MRI investigations demonstrated enhancement of left optic nerve accompanied by demyelinating lesions in supratentorial periventricular areas of occipital and temporal lobes (Table 2). Interestingly, CSF PCR analysis of SARS-CoV-2 was negative, while CSF oligoclonal IgG and IgM were positive, conferring a post-infectious immunological response as an etiology for this phenomenon [80].
An autoimmune inflammation mediated by activated microglia and cytotoxic T-cells against oligodendrocytes underlies the myelin degeneration in MS [51]. Elevated levels of pro-inflammatory cytokines (e.g., TNF-α, IL-6, IFN-γ, IL-1β) are found in the CSF of MS patients suffering from either white matter or gray matter deficits [24]. TNF-α is isolated from active areas of neurodegeneration in postmortem MS brains [57]. Overexpression of TNF-α stimulates apoptosis of oligodendrocytes and infiltration of immune cells into the CSF, while intrathecal injection of TNF-α monoclonal antibodies prevents from the development of MS in animal models of experimental autoimmune encephalomyelitis (EAE) [57]. IL-6 maintains the balance between cytotoxic and regulatory T-cells. Overproduction of IL-6 inhibits the differentiation of T-cells into regulatory T-cells and thus promotes the inflammatory state and demyelination in mice [57]. In line with this, IL-6 deficient mice are resistant to MS, and administration of IL-6 receptor (IL-6R) antibodies inhibits the myelin degeneration in EAE mice [57]. Moreover, the aberrant release of IL-6 disrupts the blood-brain barrier integrity and facilitates the infiltration of cytotoxic T-cells [57]. Last but not least, IL-1β receptor (IL-1βR) antibodies antagonize the demyelination process in rat models of EAE, which suggests a role for IL-1β in the pathogenesis of MS [81]. After all, knowing that the aforementioned cytokines surge as a result of SARS-CoV-2 mediated inflammation, we postulate that persistent inflammation in SARS-CoV-2 may propagate myelin destruction and lead to delayed progression of MS.
Moreover, it is hypothesized that an autoimmune reaction mediated by the cross-reaction between viral particles and myelin basic protein may provide the driving force for neural demyelination. This hypothesis is supported by the fact that genome of other coronaviruses like CoV-OC43 and CoV-229E, as well as their antibodies, are isolated from CNS of MS patients, and coronavirus-like particles are found in perivascular cuffs of human MS brain [82]. In fact, the virus might lie dormant in astrocytes and oligodendrocytes and trigger the autoimmunity mediated by molecular mimicry. Interestingly, intracranial inoculation of murine coronavirus (mouse hepatic virus (MHV)), induces optic neuritis and focal encephalitis leading to chronic demyelination in mice [83]. In this case, both innate and adaptive immune responses comprising of activation of microglia, B cells, CD4 T cells, and CD8 T cells along with the direct viral toxicity are proposed to mediate myelin stripping [68, 70].
Guillain-Barre Syndrome
GBS is a post-infectious autoimmune cause of polyneuropathy in predisposed individuals that leads to flaccid paralysis of lower extremities, along with autonomic and sensory deficits [69]. Autoimmunity is the milestone of GBS pathophysiology. Neurotrophic macrophages present bacterial or viral particles to lymphocytes in peripheral nervous system (PNS) [69]. The resultant immune response (i.e., complement and macrophage activation, cytokine release, and free radical production) accompanied with the molecular mimicry between pathogen epitopes and host gangliosides such as N-acetylgalactosaminyl GD1a (GalNAc-GD1a), ganglioside D1a (GD1a), ganglioside (GM1), and ganglioside M1b (GM1b) and myelin proteins result in acute neural damage [69].
GBS is reported in other post-viral infections, including influenza, enteroviruses, Zika, SARS-CoV, MERS-CoV, and SARS-CoV-2 [13]. Several cases of GBS due to SARS-CoV-2 infection are presented in the literature with the varying onset of GBS symptoms from a day before to 3 weeks after presentation of SARS-CoV-2 symptoms [71, 84]. Enhancement of cervical and cranial roots (e.g., optic, oculomotor, and facial nerves) in MRI studies along with presence of axonal and demyelinating patterns in nerve conduction studies are suggestive of GBS in SARS-CoV-2 patients (Table 2) [71, 84,85,86].
Several findings indicate that the occurrence of GBS in SARS-CoV-2 is mediated by an autoimmune response mediated by molecular mimicry and not the direct viral invasion. Binding of SARS-CoV-2 spike protein to cell surface occurs not only through ACE-2 receptors but also through sialic acid–containing glycoproteins and gangliosides. Considering the fact that anti-ganglioside antibodies are found in the serum of SARS-CoV-2-triggered GBS patients, a cross-reactivity between SARS-CoV-2-binding gangliosides and peripheral nerve glycolipids are implicated [87]. Also, recent experiments are not able to detect the viral genome in CSF of a high proportion of post-SARS-CoV-2 GBS patients [23, 39, 72]. Moreover, the administration of intravenous immunoglobulin (IVIg) resolves symptoms and improves the outcomes in post-SARS-CoV-2 GBS patients [59].
Other plausible theories may underlie the development of GBS in SARS-CoV-2. A shift in immune response toward pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α, and IFN-γ) occurs in acute stages of GBS and mediate neural degeneration [69]. Similarly, elevated levels of IL-1β and IL-6 are found in the CSF of GBS patients [69]. So, the release of pro-inflammatory cytokines in SARS-CoV-2 may propagate the neurodegeneration leading to GBS. Also, the SARS-CoV-2-mediated cytokine storm may result in disruption of blood-brain barrier integrity and infiltration of lymphocytes leading to GBS [73].
Seizure and Encephalopathy
Recent retrospective, case series, and case report studies indicate that SARS-CoV-2, as well as SARS-CoV and MERS-CoV patients, is prone to encephalopathy and seizure [88]. Brain MRI studies of these patients demonstrate multifocal involvement of cortex and white matter (including periventricular areas), along with multiple hyperintense areas in T2-weighted and FLAIR images in temporal, frontal, and occipital lobes and hippocampus (Table 2) [74, 89].
Consequences of SARS-CoV-2 infection, including organ failure, hypoxia, metabolic disturbance, and direct viral invasion into the CNS, are proposed to explain the observations of encephalopathy and seizure in these patients [88]. However, most findings are in favor of immune-mediated encephalopathy and seizure. Except for two case reports, all the remaining studies are not able to detect the viral genome in the CSF of these patients [41]. Seemingly, most patients exhibit a dramatic response to plasmapheresis and steroid therapy, which indicates a role for autoimmune-mediated injury [23]. As mentioned before, the aberrant release of pro-inflammatory cytokines like IL-1β, IL-6, and TNF-α disrupts blood-brain barrier permeability, and the resulting infiltration of WBCs and activation of microglia and astrocytes damages neurons and glial cells [24, 41]. Also, it is speculated that the viral-mediated production of autoantibodies against glial cells might be responsible for neural injury [74].
Brainstem Involvement
Transgenic mice infected by SARS-CoV and MERS-CoV exhibit brainstem involvement due to trans-synaptic projection of the virus throughout the olfactory epithelium, olfactory bulb, thalamus, and finally the brainstem [16]. The virus might target the primary respiratory oscillator (known as pre-Bötzinger complex) in the brainstem and result in respiratory failure in these animal models [16]. Seemingly, acute respiratory failure due to brainstem dysfunction is reported in SARS-CoV-2 patients [90]. Also, a recent case study reports post-infectious brainstem involvement presenting with involuntary movements, myoclonus, and hyperekplexia after gradual respiratory symptom relief in a case of SARS-CoV-2 [91]. Postmortem autopsy studies elucidate areas of hyperemia and edema and neurodegeneration in the brainstem [90]. MRI findings in a case of brainstem involvement demonstrate high signal lesions in bilateral pons in T2-FLAIR images (Table 2) [92]. These findings are in favor of brainstem involvement due to direct viral invasion. However, the absence of viral genome in CSF along with the presence of inflammatory mediators (namely TNF-α and interleukin-8 (IL-8)) and pleocytosis in CSF in a group of patients with brainstem dysfunction suggests that the aberrant immune response might underlie the brainstem dysfunction in SARS-CoV-2 [90].
Conclusion and Future Remarks
SARS-CoV-2 pandemic, beginning in December 2019, has now become a global concern. Although the acute respiratory manifestations of the disease are greatly considered, reports of neurologic sequelae are dramatically increasing. A recent cohort study of SARS-CoV-2 patients reported a 4.3% prevalence for neurologic symptoms, other than anosmia and headache [93]. Also, para-clinic investigations support the idea of post-SARS-CoV-2 CNS involvement (Table 2).
Although the CNS complications might be a consequence of direct viral invasion into the CNS, recent findings are in favor of immune-mediated CNS involvement [31]. SARS-CoV-2 genome sequences are frequently undetectable in the CSF [93]. Also, recent studies suggest that blood-brain barrier integrity is disrupted, and lymphocytic pleocytosis and WBC infiltration into the CNS occur in these patients [93]. Interestingly, the presence of auto-antibodies against gangliosides in SARS-CoV-2 patients suggests that an autoimmune phenomenon mediated by the virus might underlie the neurologic symptoms [93].
As a result, we propose that the aberrant immune response mediated by SARS-CoV-2 is responsible for the majority of acute or chronic neurological manifestations. In fact, SARS-CoV-2 infection increases the risk of Alzheimer’s disease, Parkinson’s disease, stroke, multiple sclerosis, Guillain-Barre syndrome, seizure, encephalopathy, and brainstem involvement through different immune pathways including cytokine storm, autoimmunity, microglial and inflammasome activation, gut microbiome modulation, WBC infiltration into the CNS, inflammatory-mediated vascular wall injury, and upregulation of pro-coagulative factors (Fig. 3). Precise investigation and follow-up of SARS-CoV-2 patients is warranted in this issue, as the delayed presentation of these complications may lead to underdiagnosis. Immunomodulatory drugs such as cytokine-blocking therapies (e.g., IL-6R monoclonal antibodies, TNF-α inhibitors, IL-1β antagonists), antibodies against viral spike proteins and viral binding receptors (ACE-2), anti-inflammatory drugs like nonsteroidal anti-inflammatory drug (NSAID), and prophylactic antibodies are under investigation in the prevention and treatment of SARS-CoV-2 [94, 95]. However, the efficiency of these drugs to decline the burden of short-term and long-term neurological manifestations of SARS-CoV-2 is yet to be investigated.

The pathomechanisms of immune-mediated CNS involvement in SARS-CoV-2 infection. SARS-CoV-2 induces neurodegeneration through cytokine storm, autoimmunity, microglial and inflammasome activation, and gut microbiome modulation. These pathways lead to development of Alzheimer’s disease in susceptible individuals and worsen the severity of Parkinson’s disease in affected patients. Inflammatory-mediated vascular wall injury, atrial fibrillation, carotid plaque rupture, and upregulation of pro-coagulative factors may increase the risk of stroke in these patients. Cytokine storm and autoimmunity induced by cross-reactivity between viral particles and CNS components may contribute to the development of autoimmune disorders of CNS, like Guillain-Barre syndrome. Also, the aforementioned mechanisms exacerbate symptoms in multiple sclerosis patients. Cytokine storm induces microglial activation and WBC infiltration into the CNS and, thus, precipitates seizure and encephalopathy. CNS central nervous system, WBC white blood cell. Adopted from https://www.dpz.eu/de/infothek/wissen/coronaviren.html, by Markus Hoffmann. Adopted with permission
References
Park M, Cook AR, Lim JT, Sun Y, Dickens BL (2020) A systematic review of COVID-19 epidemiology based on current evidence. J Clin Med 9(4):967
Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R (2020) Features, evaluation and treatment coronavirus (COVID-19). In: Statpearls [internet]. StatPearls Publishing,
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurology 77(6):683–690. https://doi.org/10.1001/jamaneurol.2020.1127
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, Collange O, Boulay C et al (2020) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med 382:2268–2270
Kandemirli SG, Dogan L, Sarikaya ZT, Kara S, Akinci C, Kaya D, Kaya Y, Yildirim D, Tuzuner F, Yildirim MS (2020) Brain MRI findings in patients in the intensive care unit with COVID-19 infection. Radiology:201697
Kremer S, Lersy F, de Sèze J, Ferré J-C, Maamar A, Carsin-Nicol B, Collange O, Bonneville F, Adam G, Martin-Blondel G (2020) Brain MRI findings in severe COVID-19: a retrospective observational study. Radiology:202222
Vespignani H, Colas D, Lavin BS, Soufflet C, Maillard L, Pourcher V, Paccoud O, Medjebar S et al (2020) Report of EEG finding on critically ill patients with COVID-19. Ann Neurol 88:626–630
Fischer D, Threlkeld ZD, Bodien YG, Kirsch JE, Huang SY, Schaefer PW, Rapalino O, Hochberg LR et al (2020) Intact brain network function in an unresponsive patient with COVID-19. Ann Neurol. https://doi.org/10.1002/ana.25838
Kanberg N, Ashton NJ, Andersson L-M, Yilmaz A, Lindh M, Nilsson S, Price RW, Blennow K et al (2020) Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology:10.1212/WNL.0000000000010111
Nampoothiri S, Sauve F, Ternier G, Fernandois D, Coelho C, Imbernon M, Deligia E, Perbet R, Florent V, Baroncini M (2020) The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis. bioRxiv
Archie SR, Cucullo L (2020) Cerebrovascular and neurological dysfunction under the threat of COVID-19: is there a comorbid role for smoking and vaping? Int J Mol Sci 21(11):3916
Heneka MT, Golenbock D, Latz E, Morgan D, Brown R (2020) Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res Ther 12(1):1–3
Costello F, Dalakas MC (2020) Cranial neuropathies and COVID-19: neurotropism and autoimmunity. AAN Enterprises
Umapathi T, Kor AC, Venketasubramanian N, Lim CT, Pang BC, Yeo TT, Lee CC, Lim PL et al (2004) Large artery ischaemic stroke in severe acute respiratory syndrome (SARS). J Neurol 251(10):1227–1231
Ellul M, Benjamin L, Singh B, Lant S, Michael B, Kneen R, Defres S, Sejvar J, Solomon T (2020) Neurological associations of COVID-19. Available at SSRN 3589350
Gandhi S, Srivastava AK, Ray U, Tripathi PP (2020) Is the collapse of the respiratory center in the brain responsible for respiratory breakdown in COVID-19 patients? ACS Chem Neurosci 11(10):1379–1381. https://doi.org/10.1021/acschemneuro.0c00217
Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, Liu C, Yang C (2020) Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain, behavior, and immunity
Ritchie K, Chan D, Watermeyer T (2020) The cognitive consequences of the COVID-19 epidemic: collateral damage? Brain Communications
Xia H, Lazartigues E (2008) Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem 107(6):1482–1494. https://doi.org/10.1111/j.1471-4159.2008.05723.x
Chen R, Yu J, Wang K, Howard D, French L, Chen Z, Wen C, Xu Z (2020) The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain. bioRxiv
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell
Bone RC (1996) Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit Care Med 24(1):163–172
Scoppettuolo P, Borrelli S, Naeije G (2020) Neurological involvement in SARS-CoV-2 infection: a clinical systematic review. Brain, Behavior, & Immunity-Health:100094
Serrano-Castro P, Estivill-Torrús G, Cabezudo-García P, Reyes-Bueno J, Petersen NC, Aguilar-Castillo M, Suárez-Pérez J, Jiménez-Hernández M, Moya-Molina M, Oliver-Martos B (2020) Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: a delayed pandemic? Neurología (English Edition)
Raony Í, Figueiredo CS, Pandolfo P, Giestal-De-Araujo E, Oliveira-Silva Bomfim P, Savino W (2020) Psycho-neuroendocrine-immune interactions in COVID-19: potential impacts on mental health. Front Immunol 11:1170
Yang YH, Huang YH, Chuang YH, Peng CM, Wang LC, Lin YT, Chiang BL (2005) Autoantibodies against human epithelial cells and endothelial cells after severe acute respiratory syndrome (SARS)-associated coronavirus infection. J Med Virol 77(1):1–7
Arabi Y, Harthi A, Hussein J, Bouchama A, Johani S, Hajeer A, Saeed B, Wahbi A et al (2015) Severe neurologic syndrome associated with Middle East respiratory syndrome corona virus (MERS-CoV). Infection 43(4):495–501
Troyer EA, Kohn JN, Hong S (2020) Are we facing a crashing wave of neuropsychiatric sequelae of COVID-19? Neuropsychiatric symptoms and potential immunologic mechanisms. Brain, behavior, and immunity
Kappelmann N, Lewis G, Dantzer R, Jones P, Khandaker G (2018) Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol Psychiatry 23(2):335–343
Mesci P, Macia A, Saleh A, Martin-Sancho L, Yin X, Snethlage C, Avansini S, Chanda S, Muotri A (2020) Sofosbuvir protects human brain organoids against SARS-CoV-2. bioRxiv
Panciani PP, Saraceno G, Zanin L, Renisi G, Signorini L, Battaglia L, Fontanella MM (2020) SARS-CoV-2: “three-steps” infection model and CSF diagnostic implication. Brain, Behavior, and Immunity
Feng X, Li S, Sun Q, Zhu J, Chen B, Xiong M, Cao G (2020) Immune-inflammatory parameters in COVID-19 cases: a systematic review and meta-analysis. Frontiers in Medicine 7:301
Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S (2020) Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a review. JAMA Neurology
Iyer AS, Jones FK, Nodoushani A, Kelly M, Becker M, Slater D, Mills R, Teng E, Kamruzzaman M, Garcia-Beltran WF (2020) Dynamics and significance of the antibody response to SARS-CoV-2 infection medRxiv
Xiang F, Wang X, He X, Peng Z, Yang B, Zhang J, Zhou Q, Ye H, Ma Y, Li H (2020) Antibody detection and dynamic characteristics in patients with COVID-19. Clin Infect Dis
Sinha P, Matthay MA, Calfee CS (2020) Is a “cytokine storm” relevant to COVID-19? JAMA Internal Medicine
Calsolaro V, Edison P (2016) Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimer's & dementia: the journal of the Alzheimer's Association 12(6):719–732. https://doi.org/10.1016/j.jalz.2016.02.010
Jónsdóttir HR (2016) Genetic modification of primary human airway epithelial cultures–a novel platform to study respiratory pathogens. ETH Zurich
Gutiérrez-Ortiz C, Méndez A, Rodrigo-Rey S, San Pedro-Murillo E, Bermejo-Guerrero L, Gordo-Mañas R, de Aragón-Gómez F, Benito-León J (2020) Miller Fisher Syndrome and polyneuritis cranialis in COVID-19. Neurology 95:e601–e605
Jain R, Young M, Dogra S, Kennedy H, Nguyen V, Jones S, Bilaloglu S, Hochman K, Raz E, Galetta S (2020) COVID-19 related neuroimaging findings: a signal of thromboembolic complications and a strong prognostic marker of poor patient outcome. Journal of the Neurological Sciences:116923
Fotuhi M, Mian A, Meysami S, Raji CA Neurobiology of COVID-19. Journal of Alzheimer's Disease (Preprint):1–17
Debnath M, Berk M, Maes M (2020) Changing dynamics of psychoneuroimmunology during COVID-19 pandemic. Brain, Behavior, & Immunity-Health:100096
Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S et al (2017) Gut microbiome alterations in Alzheimer's disease. Sci Rep 7(1):13537. https://doi.org/10.1038/s41598-017-13601-y
Lin CH, Chen CC, Chiang HL, Liou JM, Chang CM, Lu TP, Chuang EY, Tai YC et al (2019) Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson's disease. J Neuroinflammation 16(1):129. https://doi.org/10.1186/s12974-019-1528-y
Jacomy H, Fragoso G, Almazan G, Mushynski WE, Talbot PJ (2006) Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 349(2):335–346
Li B, Chohan MO, Grundke-Iqbal I, Iqbal K (2007) Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol 113(5):501–511
Rahmani F, Sanjari Moghaddam H, Rahmani M, Aarabi MH (2020) Metabolic connectivity in Alzheimer’s diseases. Clinical and Translational Imaging 8(3):157–166. https://doi.org/10.1007/s40336-020-00371-3
Wang H, Yuan Z, Pavel MA, Hansen SB (2020) The role of high cholesterol in age-related COVID19 lethality. bioRxiv
Kuo C-L, Pilling LC, Atkins JL, Masoli JA, Delgado J, Kuchel GA, Melzer D (2020) APOE e4 genotype predicts severe COVID-19 in the UK Biobank community cohort. medRxiv
Riedel BC, Thompson PM, Brinton RD (2016) Age, APOE and sex: triad of risk of Alzheimer's disease. J Steroid Biochem Mol Biol 160:134–147. https://doi.org/10.1016/j.jsbmb.2016.03.012
Rothaug M, Becker-Pauly C, Rose-John S (2016) The role of interleukin-6 signaling in nervous tissue. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1863(6):1218–1227
Naughton SX, Raval U, Pasinetti GM Potential novel role of COVID-19 in Alzheimer’s disease and preventative mitigation strategies. Journal of Alzheimer's Disease (Preprint):1–5
De Virgilio A, Greco A, Fabbrini G, Inghilleri M, Rizzo MI, Gallo A, Conte M, Rosato C et al (2016) Parkinson's disease: autoimmunity and neuroinflammation. Autoimmun Rev 15(10):1005–1011
Wegrzynowicz M, Bar-On D, Anichtchik O, Iovino M, Xia J, Ryazanov S, Leonov A, Giese A et al (2019) Depopulation of dense α-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson’s disease model. Acta Neuropathol 138(4):575–595
Dolatshahi M, Pourmirbabaei S, Kamalian A, Ashraf-Ganjouei A, Yaseri M, Aarabi MH (2018) Longitudinal alterations of alpha-synuclein, amyloid beta, total, and phosphorylated tau in cerebrospinal fluid and correlations between their changes in Parkinson's disease. Front Neurol 9(560). https://doi.org/10.3389/fneur.2018.00560
Sanjari Moghaddam H, Ghazi Sherbaf F, Mojtahed Zadeh M, Ashraf-Ganjouei A, Aarabi MH (2018) Association between peripheral inflammation and DATSCAN data of the striatal nuclei in different motor subtypes of Parkinson disease. Front Neurol 9(234). https://doi.org/10.3389/fneur.2018.00234
Jung YJ, Tweedie D, Scerba MT, Greig NH (2019) Neuroinflammation as a factor of neurodegenerative disease: thalidomide analogs as treatments. Frontiers in Cell and Developmental Biology 7:313
Olichney JM, Murphy C, Hofstetter C, Foster K, Hansen L, Thal L, Katzman R (2005) Anosmia is very common in the Lewy body variant of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 76(10):1342–1347
Postuma RB, Berg D (2016) Advances in markers of prodromal Parkinson disease. Nat Rev Neurol 12(11):622–634
Schapira AHV, Chaudhuri KR, Jenner P (2017) Non-motor features of Parkinson disease. Nat Rev Neurosci 18(7):435–450. https://doi.org/10.1038/nrn.2017.62
Sobhani S, Rahmani F, Aarabi MH, Sadr AV (2019) Exploring white matter microstructure and olfaction dysfunction in early parkinson disease: diffusion MRI reveals new insight. Brain imaging and behavior 13(1):210–219
Mehraeen E, Behnezhad F, Salehi MA, Noori T, Harandi H, SeyedAlinaghi S (2020) Olfactory and gustatory dysfunctions due to the coronavirus disease (COVID-19): a review of current evidence. Eur Arch Otorhinolaryngol. https://doi.org/10.1007/s00405-020-06120-6
Chung TW-H, Sridhar S, Zhang AJ, Chan K-H, Li H-L, Wong FK-C, Ng M-Y, Tsang RK-Y, Lee AC-Y, Fan Z Olfactory dysfunction in coronavirus disease 2019 patients: observational cohort study and systematic review. In: Open forum infectious diseases, 2020. vol 6. Oxford University Press US, p ofaa199
Pellegrino R, Cooper KW, Di Pizio A, Joseph PV, Bhutani S, Parma V (2020) Coronaviruses and the chemical senses: past, present, and future. COVID-19 Research
Bilinska K, Jakubowska P, Von Bartheld CS, Butowt R (2020) Expression of the SARS-CoV-2 entry proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: identification of cell types and trends with age. ACS Chem Neurosci 11:1555–1562
Meinhardt J, Radke J, Dittmayer C, Mothes R, Franz J, Laue M, Schneider J, Bruenink S, Hassan O, Stenzel W (2020) Olfactory transmucosal SARS-CoV-2 invasion as port of central nervous system entry in COVID-19 patients. bioRxiv
Karimi-Galougahi M, Yousefi-Koma A, Bakhshayeshkaram M, Raad N, Haseli S (2020) 18FDG PET/CT scan reveals hypoactive orbitofrontal cortex in anosmia of COVID-19. Acad Radiol 27:1042–1043
Kishore A, Kanaujia A, Nag S, Rostami AM, Kenyon LC, Shindler KS, Das Sarma J (2013) Different mechanisms of inflammation induced in virus and autoimmune-mediated models of multiple sclerosis in C57BL6 mice. Biomed Res Int 2013:589048–589049. https://doi.org/10.1155/2013/589048
Soltani ZE, Rahmani F, Rezaei N (2019) Autoimmunity and cytokines in Guillain-Barré syndrome revisited: review of pathomechanisms with an eye on therapeutic options. Eur Cytokine Netw 30(1):1–14
Bender SJ, Weiss SR (2010) Pathogenesis of murine coronavirus in the central nervous system. J Neuroimmune Pharmacol 5(3):336–354. https://doi.org/10.1007/s11481-010-9202-2
Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, Jayaseelan DL, Kumar G, Raftopoulos RE, Zambreanu L (2020) The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain
Pryce-Roberts A, Talaei M, Robertson N (2020) Neurological complications of COVID-19: a preliminary review. Journal of Neurology:1
Zhou Z, Kang H, Li S, Zhao X (2020) Understanding the neurotropic characteristics of SARS-CoV-2: from neurological manifestations of COVID-19 to potential neurotropic mechanisms. Journal of Neurology:1
Zanin L, Saraceno G, Panciani PP, Renisi G, Signorini L, Migliorati K, Fontanella MM (2020) SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir:1–4
Goldberg MF, Goldberg MF, Cerejo R, Tayal A (2020) Cerebrovascular disease in COVID-19. Am J Neuroradiol 41:1170–1172
Altable M, de la Serna JM Cerebrovascular Disease in COVID-19: Is there a higher risk of stroke?
Ghannam M, Alshaer Q, Al-Chalabi M, Zakarna L, Robertson J, Manousakis G (2020) Neurological involvement of coronavirus disease 2019: a systematic review.
Lambertsen KL, Biber K, Finsen B (2012) Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 32(9):1677–1698
Kim JS (2020) Coronavirus disease 2019 and stroke. Journal of Stroke 22(2):157–158
Palao M, Fernández-Díaz E, Gracia-Gil J, Romero-Sánchez C, Díaz-Maroto I, Segura T (2020) Multiple sclerosis following SARS-CoV-2 infection. Multiple sclerosis and related disorders:102377
Wang K, Song F, Fernandez-Escobar A, Luo G, Wang J-H, Sun Y (2018) The properties of cytokines in multiple sclerosis: pros and cons. Am J Med Sci 356(6):552–560
Montalvan V, Lee J, Bueso T, De Toledo J, Rivas K (2020) Neurological manifestations of COVID-19 and other coronavirus infections: a systematic review. Clin Neurol Neurosurg 194:105921
Shindler KS, Kenyon LC, Dutt M, Hingley ST, Sarma JD (2008) Experimental optic neuritis induced by a demyelinating strain of mouse hepatitis virus. J Virol 82(17):8882–8886
Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, Franciotta D, Baldanti F et al (2020) Guillain–Barré syndrome associated with SARS-CoV-2. N Engl J Med 382:2574–2576
Dinkin M, Gao V, Kahan J, Bobker S, Simonetto M, Wechsler P, Harpe J, Greer C et al (2020) COVID-19 presenting with ophthalmoparesis from cranial nerve palsy. Neurology 95:221–223
Lantos J, Strauss S, Lin E (2020) COVID-19–associated miller fisher syndrome: MRI findings. Am J Neuroradiol 41:1184–1186
Dalakas MC (2020) Guillain-Barré syndrome: the first documented COVID-19–triggered autoimmune neurologic disease: more to come with myositis in the offing. Neurology-Neuroimmunology Neuroinflammation 7(5):e781
Asadi-Pooya AA (2020) Seizures associated with coronavirus infections. Seizure 79:49–52
Anzalone N, Castellano A, Scotti R, Scandroglio AM, Filippi M, Ciceri F, Tresoldi M, Falini A (2020) Multifocal laminar cortical brain lesions: a consistent MRI finding in neuro-COVID-19 patients. J Neurol 1
Jasti M, Nalleballe K, Dandu V, Onteddu S (2020) A review of pathophysiology and neuropsychiatric manifestations of COVID-19. J Neurol 1
Khoo A, McLoughlin B, Cheema S, Weil RS, Lambert C, Manji H, Zandi MS, Morrow JM (2020) Postinfectious brainstem encephalitis associated with SARS-CoV-2. Journal of Neurology, Neurosurgery & Psychiatry
Afshar H, Yassin Z, Kalantari S, Aloosh O, Lotfi T, Moghaddasi M, Sadeghipour A, Emamikhah M (2020) Evolution and resolution of brain involvement associated with SARS-CoV2 infection: a close clinical–paraclinical follow up study of a case. Multiple Sclerosis and Related Disorders 102216
Guilmot A, Slootjes SM, Sellimi A, Bronchain M, Hanseeuw B, Belkhir L, Yombi JC, De Greef J et al (2020) Immune-mediated neurological syndromes in SARS-CoV-2-infected patients. J Neurol:1–7
Zabetakis I, Lordan R, Norton C, Tsoupras A (2020) COVID-19: The inflammation link and the role of nutrition in potential mitigation. Nutrients 12(5):1466
Bhatti JS, Bhatti GK, Khullar N, Reddy AP, Reddy PH (2020) Therapeutic strategies in the development of anti-viral drugs and vaccines against SARS-CoV-2 infection. Mol Neurobiol. https://doi.org/10.1007/s12035-020-02074-2
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mohammadi, S., Moosaie, F. & Aarabi, M.H. Understanding the Immunologic Characteristics of Neurologic Manifestations of SARS-CoV-2 and Potential Immunological Mechanisms. Mol Neurobiol 57, 5263–5275 (2020). https://doi.org/10.1007/s12035-020-02094-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02094-y