
Abstract
Cerebrospinal fluid (CSF) total prion protein (t-PrP) is decreased in sporadic Creutzfeldt-Jakob disease (sCJD). However, data on the comparative signatures of t-PrP across the spectrum of prion diseases, longitudinal changes during disease progression, and levels in pre-clinical cases are scarce. T-PrP was quantified in neurological diseases (ND, n = 147) and in prion diseases from different aetiologies including sporadic (sCJD, n = 193), iatrogenic (iCJD, n = 12) and genetic (n = 209) forms. T-PrP was also measured in serial lumbar punctures obtained from sCJD cases at different symptomatic disease stages, and in asymptomatic prion protein gene (PRNP) mutation carriers. Compared to ND, t-PrP concentrations were significantly decreased in sCJD, iCJD and in genetic prion diseases associated with the three most common mutations E200K, V210I (associated with genetic CJD) and D178N-129M (associated with fatal familial insomnia). In contrast, t-PrP concentrations in P102L mutants (associated with the Gerstmann-Sträussler-Scheinker syndrome) remained unaltered. In serial lumbar punctures obtained at different disease stages of sCJD patients, t-PrP concentrations inversely correlated with disease progression. Decreased mean t-PrP values were detected in asymptomatic D178-129M mutant carriers, but not in E200K and P102L carriers. The presence of low CSF t-PrP is common to all types of prion diseases regardless of their aetiology albeit with mutation-specific exceptions in a minority of genetic cases. In some genetic prion disease, decreased levels are already detected at pre-clinical stages and diminish in parallel with disease progression. Our data indicate that CSF t-PrP concentrations may have a role as a pre-clinical or early symptomatic diagnostic biomarker in prion diseases as well as in the evaluation of therapeutic interventions.





Similar content being viewed by others
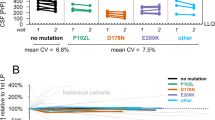
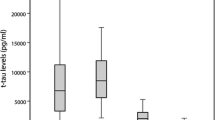

Abbreviations
- GSS-S:
-
Gerstmann–Sträussler–Scheinker syndrome
- ROC:
-
Receiver operating characteristic
- OPRI:
-
Octapeptide repeat insertion
- PRNP:
-
Prion protein gene
- gCJD:
-
Genetic Creutzfeldt-Jakob disease
- iCJD:
-
Iatrogenic Creutzfeldt-Jakob disease
- vCJD:
-
Variant Creutzfeldt-Jakob disease
- FFI:
-
Fatal Familial Insomnia
- NFL:
-
Neurofilament light
- PrPscz:
-
PrPsc: Prion protein scrapie
- LP:
-
Lumbar puncture
- AUC:
-
Area under the curve
- sCJD:
-
Sporadic Creutzfeldt-Jakob disease
- CSF:
-
Cerebrospinal fluid
- ELISA:
-
Enzyme-linked immunosorbent assays
- ND:
-
Neurological diseases
- t-PrP:
-
Total prion protein
References
Aguzzi A, Sigurdson C, Heikenwaelder M (2008) Molecular mechanisms of prion pathogenesis. Annu Rev Pathol Mech Dis 3:11–40
Aguzzi A (2006) Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. J Neurochem 97:1726–1739
Chen C, Dong X-P (2016) Epidemiological characteristics of human prion diseases. Infect Dis Poverty [Internet] 5:47. Available from: http://idpjournal.biomedcentral.com/articles/10.1186/s40249-016-0143-8
Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG et al (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol [Internet] 39:767–78. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8651649
Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O et al (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233
Mastrianni JA (2010) The genetics of prion diseases. Genet Med 12:187–95
Lloyd SE, Mead S, Collinge J (2013) Genetics of prion diseases. Curr Opin Genet Dev 345–51
Bonda DJ, Manjila S, Mehndiratta P, Khan F, Miller BR, Onwuzulike K et al (2016) Human prion diseases: surgical lessons learned from iatrogenic prion transmission. Neurosurg Focus 41:E10
Zerr I, Bodemer M, Gefeller O, Otto M, Poser S, Wiltfang J, Windl O, Kretzschmar HA et al (1998) Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann Neurol 43:32–40
Otto M, Wiltfang J, Tumani H, Zerr I, Lantsch M, Kornhuber J et al (1997) Elevated levels of tau-protein in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Neurosci Lett 225:210–212
Llorens F, Kruse N, Schmitz M, Gotzmann N, Golanska E, Thüne K, Zejneli O, Kanata E et al (2017) Evaluation of α-synuclein as a novel cerebrospinal fluid biomarker in different forms of prion diseases. Alzheimers Dement 13:710–719
Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, Matsubara T, Nakagaki T et al (2011) Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 17:175–178
Meyne F, Gloeckner SF, Ciesielczyk B, Heinemann U, Krasnianski A, Meissner B, Zerr I (2009) Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J Alzheimers Dis 17:863–873
Dorey A, Tholance Y, Vighetto A, Perret-Liaudet A, Lachman I, Krolak-Salmon P, Wagner U, Struyfs H et al (2015) Association of cerebrospinal fluid prion protein levels and the distinction between Alzheimer disease and Creutzfeldt-Jakob disease. JAMA Neurol 72:267–275
Rumeileh SA, Lattanzio F, Maserati MS, Rizzi R, Capellari S, Parchi P (2016) Diagnostic accuracy of a combined analysis of cerebrospinal fluid t-PrP, t-tau, p-tau, and Aβ42 in the differential diagnosis of Creutzfeldt-Jakob disease from Alzheimer’s disease with emphasis on atypical disease variants. J Alzheimers Dis 55:1–10
Llorens F, Ansoleaga B, Garcia-Esparcia P, Zafar S, Grau-Rivera O, López-González I, Blanco R et al (2013) PrP mRNA and protein expression in brain and PrP(c) in CSF in Creutzfeldt-Jakob disease MM1 and VV2. Prion 7:383–93
Schmitz M, Schlomm M, Hasan B, Beekes M, Mitrova E, Korth C, Breil A, Carimalo J et al (2010) Codon 129 polymorphism and the E200K mutation do not affect the cellular prion protein isoform composition in the cerebrospinal fluid from patients with Creutzfeldt-Jakob disease. Eur J Neurosci 31:2024–2031
Torres M, Cartier L, Matamala JM, Hernández N, Woehlbier U, Hetz C (2012) Altered prion protein expression pattern in CSF as a biomarker for Creutzfeldt-Jakob disease. PLoS One 7:e36159
Schmitz M, Lüllmann K, Zafar S, Ebert E, Wohlhage M, Oikonomou P, Schlomm M, Mitrova E et al (2014) Association of prion protein genotype and scrapie prion protein type with cellular prion protein charge isoform profiles in cerebrospinal fluid of humans with sporadic or familial prion diseases. Neurobiol Aging 35:1177–1188
Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, Breithaupt M, Varges D et al (2009) Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 132:2659–2668
Parchi P, De Boni L, Saverioni D, Cohen ML, Ferrer I, Gambetti P et al (2012) Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol 124:517–529
Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A et al (2005) Genetic prion disease: the EUROCJD experience. Hum Genet 118:166–174
World Health Organisation (2003) WHO manual for surveillance of human transmissible spongiform encephalopathies including variant Creutzfeldt-Jakob disease. WHO Man. Surveill. Hum. Transm. spongiform Enceph 105
Windl O, Giese A, Schulz-Schaeffer W, Zerr I, Skworc K, Arendt S, Oberdieck C, Bodemer M et al (1999) Molecular genetics of human prion diseases in Germany. Hum Genet 105:244–252
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Müller M (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12:77. https://doi.org/10.1186/1471-2105-12-77
Llorens F, Schmitz M, Karch A, Cramm M, Lange P, Gherib K, Varges D, Schmidt C et al (2016) Comparative analysis of cerebrospinal fluid biomarkers in the differential diagnosis of neurodegenerative dementia. Alzheimers Dement 12:577–589
Ladogana A, Sanchez-Juan P, Mitrova E, Green A, Cuadrado-Corrales N, Sanchez-Valle R et al (2009) Cerebrospinal fluid biomarkers in human genetic transmissible spongiform encephalopathies. J Neuro 256:1620–8
Cramm M, Schmitz M, Karch A, Mitrova E, Kuhn F, Schroeder B, Raeber A, Varges D et al (2016) Stability and reproducibility underscore utility of RT-QuIC for diagnosis of Creutzfeldt-Jakob disease. Mol Neurobiol 53:1896–1904
Croes EA, Theuns J, Houwing-Duistermaat JJ, Dermaut B, Sleegers K, Roks G, van den Broeck M, van Harten B et al (2004) Octapeptide repeat insertions in the prion protein gene and early onset dementia. J Neurol Neurosurg Psychiatry 75:1166–1170
Kovács GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RSG, Budka H (2002) Mutations of the prion protein gene: phenotypic spectrum. J Neurol 249:1567–1582
Schmitz M, Dittmar K, Llorens F, Gelpi E, Ferrer I, Schulz-Schaeffer WJ et al (2016) Hereditary human prion diseases: an update. Mol Neurobiol 54:4138–4149
Beck JA, Poulter M, Campbell TA, Adamson G, Uphill JB, Guerreiro R, Jackson GS, Stevens JC, Manji H, Collinge J, Mead S (2010) PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat 31(7):E1551–15563. https://doi.org/10.1002/humu.21281
Montagna P, Cortelli P, Avoni P, Tinuper P, Plazzi G, Gallassi R et al (1998) Clinical features of fatal familial insomnia: phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathol [Internet] 8:515–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9669701
Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003) Sporadic and familial CJD: classification and characterisation. Br Med Bull 66:213–239
Sanchez-Juan P, Sánchez-Valle R, Green A, Ladogana A, Cuadrado-Corrales N, Mitrová E, Stoeck K, Sklaviadis T et al (2007) Influence of timing on CSF tests value for Creutzfeldt-Jakob disease diagnosis. J Neurol 254:901–906
Llorens F, Kruse N, Karch A, Schmitz M, Zafar S, Gotzmann N, Sun T, Köchy S et al (2018) Validation of α-synuclein as a CSF biomarker for sporadic Creutzfeldt-Jakob disease. Mol Neurobiol Mol Neurobiol 55:2249–2257
Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF, McLean CY, Tung JY et al (2016) Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med 8:322ra9
Parchi P, Castellani R, Cortelli P, Montagna P, Chen SG, Petersen RB, Manetto V, Vnencak-Jones CL et al (1995) Regional distribution of protease-resistant prion protein in fatal familial insomnia. Ann Neurol 38:21–29
Llorens F, Karch A, Golanska E, Schmitz M, Lange P, Sikorska B et al (2017) Cerebrospinal fluid biomarker-based diagnosis of sporadic Creutzfeldt-Jakob disease: a validation study for previously established cutoffs. Dement Geriatr Cogn Disord 43:71–80
Zerr I, Schmitz M, Karch A, Villar-Piqué A, Kanata E, Golanska E et al (2018) Cerebrospinal fluid neurofilament light levels in neurodegenerative dementia: evaluation of diagnostic accuracy in the differential diagnosis of prion diseases. Alzheimer’s Dement 14:751–763
Llorens F, Thüne K, Schmitz M, Ansoleaga B, Frau-Méndez MA, Cramm M et al (2016) Identification of new molecular alterations in fatal familial insomnia. Hum Mol Genet 25:2417–2436
Llorens F, Barrio T, Correia Â, Villar-Piqué A, Thüne K, Lange P et al (2018) Cerebrospinal fluid prion disease biomarkers in pre-clinical and clinical naturally occurring scrapie. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1014-z
Acknowledgements
We thank Silja Köchy for indispensable technical assistance.
Funding
This study was funded by Robert Koch Institute through funds from the Federal Ministry of Health of Germany (grant no. 1369–341) to IZ, by the Spanish Ministry of Health - Instituto Carlos III/ Fondo Social Europeo (CP16/00041) to FL. This project has been funded at 65% by the Fondo Europeo de Desarrollo Regional (FEDER) through the Interreg V-A España-Francia-Andorra (POCTEFA 2014-2020) programme.
Author information
Authors and Affiliations
Contributions
AV-P, MS, IZ and FL designed the study. AV-P, MS and FL performed experiments. AV-P, MS, AK, IZ and FL analysed data and interpreted the results. IL, OC, CS, SS, AL, AP, IS, IF, EM, D.Z, MP, IB, MC, SJC, MDG, RS-V and IZ contributed to samples and/or technical expertise. FL and AV-P wrote the manuscript draft. All authors critically revised the manuscript and approved its content before submission.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
The study was conducted according to the revised Declaration of Helsinki and Good Clinical Practice guidelines, and was approved by all local Ethics committees. All study participants or their legal guardians provided written informed consent.
Consent for Publication
Not applicable.
Competing Interests
Dr. Lachmann reports he is a representative of AJ Roboscreen GmbH, Leipzig, Germany.
Additional information
Inga Zerr and Franc Llorens are equal senior contributors.
Electronic Supplementary Material
Supplementary Figure 1
CSF t-PrP levels in gPD associated to E200K, D178N-M and P102L mutations in different cohorts. (A) CSF t-PrP in E200K cases from four cohorts. (B) CSF t-PrP in D178N-M cases from two cohorts. (C) CSF t-PrP in V210I cases from two cohorts. Kruskal-Wallis test followed by Dunn’s post-hoc tests (correction for multiple testing) was applied for multiple comparisons and Mann-Whitney-U test for two group comparisons. No statistical differences were detected for any of the comparisons. (PNG 97 kb)
Supplementary Figure 2
CSF t-PrP concentrations in asymptomatic PRNP mutant carriers from the UCSF cohort (asymptomatic – UCSF), symptomatic cases from the UCSF cohort (symptomatic – UCSF) and symptomatic cases from all the cases analyzed in the present study (symptomatic - ALL) for the E200K, D178N-M and P102L mutations. Dashed red lines indicate upper and lower 95% CI t-PrP concentrations in ND cases. (PNG 99 kb)
Rights and permissions
About this article

Cite this article
Villar-Piqué, A., Schmitz, M., Lachmann, I. et al. Cerebrospinal Fluid Total Prion Protein in the Spectrum of Prion Diseases. Mol Neurobiol 56, 2811–2821 (2019). https://doi.org/10.1007/s12035-018-1251-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1251-1