
Abstract
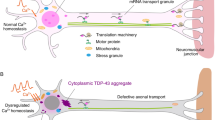
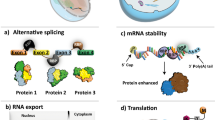
Motor neuron diseases are neurodegenerative disorders that trigger motor neurons to degenerate and lead to paralysis. Our understanding on the mechanisms of motor neuron degeneration has been enhanced by recent cellular and molecular researches. In this review, I highlight advances in RNA-binding proteins associated molecular mechanisms of motor neuron degeneration pathogenesis including ribonucleoprotein-associated motor neuron degeneration which is focused on RNA-binding protein aggregation associated aberrant RNA metabolism and stress granules mediated translational repression, neurofilament associated aggregate formation, and prion protein associated accumulation of misfolded proteins. Progress overviewed above will be valuable to the development of more targeted diagnostic tests and therapies.

Similar content being viewed by others

References
Ibrahim, F., Nakaya, T., & Mourelatos, Z. (2012). RNA dysregulation in diseases of motor neurons. Annual Review of Pathology: Mechanism, 7, 323–352.
Pandya, R. S., Zhu, H. N., Li, W., Bowser, R., Friedlander, R. M., & Wang, X. (2013). Therapeutic neuroprotective agents for amyotrophic lateral sclerosis. Cellular and Molecular Life Sciences, 70, 4729–4745.
Liu, Q., Xie, F., Siedlak, S. L., Nunomura, A., Honda, K., Moreira, P. I., et al. (2004). Neurofilament proteins in neurodegenerative diseases. Cellular and Molecular Life Science, 61, 3057–3075.
Teuling, E., Ahmed, S., Haasdijk, E., Demmers, J., Steinmetz, M. O., Akhmanova, A., et al. (2007). Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. Journal of Neuroscience, 27, 9801–9815.
Carmeliet, P., & de Almodovar, C. R. (2013). VEGF ligands and receptors: Implications in neurodevelopment and neurodegeneration. Cellular and Molecular Life Science, 70, 1763–1778.
Nizzardo, M., Simone, C., Falcone, M., Riboldi, G., Rizzo, F., Magri, F., et al. (2012). Research advances in gene therapy approaches for the treatment of amyotrophic lateral sclerosis. Cellular and Molecular Life Science, 69, 1641–1650.
Morrison, B. E., Majdzadeh, N., & D’Mello, S. R. (2007). Histone deacetylases: Focus on the nervous system. Cellular and Molecular Life Science, 64, 2258–2269.
Valori, C. F., Brambilla, L., Martorana, F., & Rossi, D. (2014). The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cellular and Molecular Life Science, 71, 287–297.
Miller, C. C. J., Ackerley, S., Brownlees, J., Grierson, A. J., Jacobsen, N. J. O., & Thornhill, P. (2002). Axonal transport of neurofilaments in normal and disease states. Cellular and Molecular Life Science, 59, 323–330.
Philips, A. V., & Cooper, T. A. (2000). RNA processing and human disease. Cellular and Molecular Life Science, 57, 235–249.
Ravindranath, R. M. H., Ravindranath, M. H., & Graves, M. C. (1997). Augmentation of natural antiganglioside IgM antibodies in lower motor neuron disease (LMND) and role of CD5+ B cells. Cellular and Molecular Life Science, 53, 750–758.
Souii, A., Ben M’hadheb-Gharbi, M., & Gharbi, J. (2013). Role of RNA structure motifs in IRES-dependent translation initiation of the coxsackievirus B3: New insights for developing live-attenuated strains for vaccines and gene therapy. Molecular Biotechnology, 55, 179–202.
Al-Fageeh, M. B., & Smales, C. M. (2013). Alternative promoters regulate cold inducible RNA-binding (CIRP) gene expression and enhance transgene expression in mammalian cells. Molecular Biotechnology, 54, 238–249.
Baumann, M., Pontiller, J., & Ernst, W. (2010). Structure and basal transcription complex of RNA polymerase II core promoters in the mammalian genome: An overview. Molecular Biotechnology, 45, 241–247.
Buratti, E., Romano, M., & Baralle, F. E. (2013). TDP-43 high throughput screening analyses in neurodegeneration: Advantages and pitfalls. Molecular and Cellular Neuroscience, 56, 465–474.
Musunuru, K. (2003). Cell-specific RNA-binding proteins in human disease. Trends in Cardiovascular Medicine, 13, 188–195.
Shi, J., Wang, Q., Johansson, J. U., Liang, X., Woodling, N. S., Priyam, P., et al. (2012). Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Annals of Neurology, 72, 788–798.
Zhang, T., Hwang, H. Y., Hao, H., Talbot, C, Jr, & Wang, J. (2012). Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. The Journal of Biological Chemistry, 287, 8371–8382.
Ghazi-Noori, S., Froud, K. E., Mizielinska, S., Powell, C., Smidak, M., Fernandez de Marco, M., et al. (2012). Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain: A Journal of Neurology, 135, 819–832.
Lopez de Maturana, R., Aguila, J. C., Sousa, A., Vazquez, N., Del Rio, P., Aiastui, A., et al. (2014). Leucine-rich repeat kinase 2 modulates cyclooxygenase 2 and the inflammatory response in idiopathic and genetic Parkinson’s disease. Neurobiology of Aging, 35, 1116–1124.
Blokhuis, A. M., Groen, E. J. N., Koppers, M., van den Berg, L. H., & Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathologia, 125, 777–794.
Van Damme, P., & Robberecht, W. (2009). Recent advances in motor neuron disease. Current Opinion in Neurology, 22, 486–492.
Neumann, M., Igaz, L. M., Kwong, L. K., Nakashima-Yasuda, H., Kolb, S. J., Dreyfuss, G., et al. (2007). Absence of heterogeneous nuclear ribonucleoproteins and survival motor neuron protein in TDP-43 positive inclusions in frontotemporal lobar degeneration. Acta Neuropathologia, 113, 543–548.
Budini, M., Baralle, F. E., & Buratti, E. (2011). Regulation of gene expression by TDP-43 and FUS/TLS in frontotemporal lobar degeneration. Current Alzheimer Research, 8, 237–245.
Fiesel, F. C., & Kahle, P. J. (2011). TDP-43 and FUS/TLS: Cellular functions and implications for neurodegeneration. FEBS Journal, 278, 3550–3568.
Baloh, R. H. (2011). TDP-43: The relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. FEBS Journal, 278, 3539–3549.
Ule, J. (2008). Ribonucleoprotein complexes in neurologic diseases. Current Opinion in Neurobiology, 18, 516–523.
Bentmann, E., Haass, C., & Dormann, D. (2013). Stress granules in neurodegeneration: Lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS Journal, 280, 4348–4370.
Luong, K. V. Q., & Nguyen, L. T. H. (2013). Roles of vitamin D in amyotrophic lateral sclerosis: Possible genetic and cellular signaling mechanisms. Molecular Brain, 6, 16.
Burman, J. L., Bourbonniere, L., Philie, J., Stroh, T., Dejgaard, S. Y., Presley, J. F., et al. (2008). Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. Journal of Biological Chemistry, 283, 22774–22786.
Oh, Y. K., Shin, K. S., Yuan, J. Y., & Kang, S. J. (2008). Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. Journal of Neurochemistry, 104, 993–1005.
Romano, M., & Buratti, E. (2013). Targeting RNA binding proteins involved in neurodegeneration. Journal of Biomolecular Screen, 18, 967–983.
Lee, E. B., Lee, V. M. Y., & Trojanowski, J. Q. (2012). Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nature Review Neuroscience, 13, 38–50.
Dewey, C. M., Cenik, B., Sephton, C. F., Johnson, B. A., Herz, J., & Yu, G. (2012). TDP-43 aggregation in neurodegeneration: Are stress granules the key? Brain Research, 1462, 16–25.
Polymenidou, M., & Cleveland, D. W. (2011). The seeds of neurodegeneration: Prion-like spreading in ALS. Cell, 147, 498–508.
Strong, M. J. (2010). The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). Journal of Neurological Science, 288, 1–12.
Lanson, N. A., & Pandey, U. B. (2012). FUS-related proteinopathies: Lessons from animal models. Brain Research, 1462, 44–60.
Miyazaki, K., Yamashita, T., Morimoto, N., Sato, K., Mimoto, T., Kurata, T., et al. (2013). Early and selective reduction of NOP56 (Asidan) and RNA processing proteins in the motor neuron of ALS model mice. Neurological Research, 35, 744–754.
Volkening, K., Leystra-Lantz, C., Yang, W. C., Jaffee, H., & Strong, M. J. (2009). Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Research, 1305, 168–182.
Fujita, K., Ito, H., Nakano, S., Kinoshita, Y., Wate, R., & Kusaka, H. (2008). Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathologia, 116, 439–445.
Elliott, J. L., & Snider, W. D. (1995). Parvalbumin is a marker of Als-resistant motor-neurons. NeuroReport, 6, 449–452.
Szaro, B. G., & Strong, M. J. (2010). Post-transcriptional control of neurofilaments: New roles in development, regeneration and neurodegenerative disease. Trends in Neuroscience, 33, 27–37.
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature, 495, 467–472.
Kanekura, K., Nishimoto, I., Aiso, S., & Matsuoka, M. (2006). Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). Journal of Biological Chemistry, 281, 30223–30233.
Blokhuis, A. M., Groen, E. J., Koppers, M., van den Berg, L. H., & Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathologica, 125, 777–794.
Ayers, J. I., Kincaid, A. E., & Bartz, J. C. (2009). Prion strain targeting independent of strain-specific neuronal tropism. Journal of Virology, 83, 81–87.
Worrall, B. B., Rowland, L. P., Chin, S. S., & Mastrianni, J. A. (2000). Amyotrophy in prion diseases. Archives of Neurology, 57, 33–38.
Talbot, K. (2014). Amyotrophic lateral sclerosis: Cell vulnerability or system vulnerability? Journal of Anatomy, 224, 45–51.
Grad, L.I., & Cashman, N.R. (2014). Prion-like activity of Cu/Zn superoxide dismutase: Implications for amyotrophic lateral sclerosis. Prion, 8.
Kishimoto, Y., Hirono, M., Atarashi, R., Sakaguchi, S., Yoshioka, T., Katamine, S., et al. (2013). Age-dependent impairment of eyeblink conditioning in prion protein-deficient mice. PLoS One, 8, e60627.
King, O. D., Gitler, A. D., & Shorter, J. (2012). The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Research, 1462, 61–80.
Senatore, A., Colleoni, S., Verderio, C., Restelli, E., Morini, R., Condliffe, S. B., et al. (2012). Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC alpha(2)delta-1 subunit. Neuron, 74, 300–313.
Gitler, A. D., & Shorter, J. (2011). RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion, 5, 179–187.
Festoff, B. M., D’Andrea, M. R., Citron, B. A., Salcedo, R. M., Smirnova, I. V., & Andrade-Gordon, P. (2000). Motor neuron cell death in wobbler mutant mice follows overexpression of the G-protein-coupled, protease-activated receptor for thrombin. Molecular Medicine, 6, 410–429.
Jokic, N., Ling, Y. Y., Ward, R. E., Michael-Titus, A. T., Priestley, J. V., & Malaspina, A. (2007). Retinoid receptors in chronic degeneration of the spinal cord: Observations in a rat model of amyotrophic lateral sclerosis. Journal of Neurochemistry, 103, 1821–1833.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tang, A.Y. RNA-Binding Proteins Associated Molecular Mechanisms of Motor Neuron Degeneration Pathogenesis. Mol Biotechnol 56, 779–786 (2014). https://doi.org/10.1007/s12033-014-9785-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-014-9785-6