
Abstract
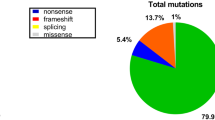
This manuscript aimed to determine the underlying point mutations causing Duchenne muscular dystrophy (DMD) in a heterogeneous group of Iranian patients, who are clinically suspected. Whole-exome sequencing was utilized to detect disease-causing variants in 40 MLPA-negative DMD patients. Disease-causing variants were detected in the DMD gene in 36/40 of the patients (90%), and 4/40 of them (10%) remained undiagnosed. WES analysis revealed that nonsense variant was the most common type in our study (23/36 of the cases). Besides, 12/36 of the cases had frameshift variant, and one of the patients had a likely pathogenic splice variant in the DMD gene. Carrier testing revealed that 21/40 of the mothers had the identified variant. Therefore, most variants were inherited (58.3%), while 19/40 were de novo (41. 7%). The present study has demonstrated the importance of performing WES to detect disease-causing point mutations in MLPA-negative DMD patients and to identify carrier females. Due to regulatory challenges, the clinical development of therapeutic approaches is time-consuming and may not be available to all patients shortly. Therefore, it appears that the techniques used to accurately detect disease-causing variants in carrier mothers are a more efficient solution to prevent the increased prevalence of DMD.



Similar content being viewed by others


Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Barzegar M, Habibi P, Bonyady M, Topchizadeh V, Shiva S (2015) Exon deletion pattern in Duchene muscular dystrophy in north west of Iran. Iranian J Child Neurol 9:42–48
Berger J, Li M, Berger S, Meilak M, Rientjes J, Currie P, Medicine M (2020) Effect of Ataluren on dystrophin mutations. J Cell Mol Med 24:6680–6689
Campbell C, Barohn RJ, Bertini E, Chabrol B, Comi GP, Darras BT, Finkel RS, Flanigan KM, Goemans N, Iannaccone ST, Jones KJ, Kirschner J, Mah JK, Mathews KD, McDonald CM, Mercuri C, Nevo Y, Péréon Y, Renfroe JB, Ryan MM, Sampson JB, Schara U, Sejersen T, Selby K, Tulinius M, Vílchez JJ, Voit T, Wei LJ, Wong BL, Elfring G, Souza M, McIntosh J, Trifillis P, Peltz SW, Muntoni F (2020) Meta-analyses of Ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. Future Med 9:973–984
Cho A, Seong MW, Lim BC, Lee HJ, Byeon JH, Kim SS, Kim SY, Choi SA, Wong AL, Lee HJ (2017) Consecutive analysis of mutation spectrum in the dystrophin gene of 507 Korean boys with Duchenne/Becker muscular dystrophy in a single center. Muscle Nerve 55:727–734
Daoud F, Angeard N, Demerre B, Martie I, Benyaou R, Leturcq F, Cossée M, Deburgrave N, Saillour Y, Tuffery S (2009) Analysis of Dp71 contribution in the severity of mental retardation through comparison of Duchenne and Becker patients differing by mutation consequences on Dp71 expression. Hum Mol Genet 18:3779–3794
Hoffman EP (2020) The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J 287:3879–3887
Jankowski S, Currie-Fraser E, Xu L, Coffa J (2008) Multiplex ligation-dependent probe amplification analysis on capillary electrophoresis instruments for a rapid gene copy number study. J Biomol Tech 19:238–243
Koeks Z, Bladen CL, Salgado D, Van Zwet E, Pogoryelova O, Mcmacken G, Monges S, Foncuberta ME, Kekou K, Kosma K (2017) Clinical outcomes in Duchenne muscular dystrophy: a study of 5345 patients from the TREAT-NMD DMD global database. IOS Press J 4:293–306
Kumar SH, Athimoolam K, Suraj M, Das Christu Das MS, Muralidharan A, Jeyam D, Ashokan J, Karthikeyan P, Krishna R, Khanna-Gupta A, Raman BL (2020) Comprehensive genetic analysis of 961 unrelated Duchenne muscular dystrophy patients: focus on diagnosis, prevention and therapeutic possibilities. PLoS One 15:e0232654
Li Y, Liu Z, Ouyang S, Zhu Y, Wang L, Wu J (2016) Distribution of dystrophin gene deletions in a Chinese population. J Int Med Res 44:99–108
López-Ferrando V, Gazzo A, De La Cruz X, Orozco M, Gelpí JL (2017) PMut: a web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res 45:W222–W228
Luce LN, Carcione M, Mazzanti C, Ferrer M, Szijan I, Giliberto F (2018) Small mutation screening in the DMD gene by whole exome sequencing of an argentine Duchenne/Becker muscular dystrophies cohort. Neuromuscul Disord 28:986–995
Lucena-Aguilar G, Sánchez-López AM, Barberán-Aceituno C, Carrillo-Avila JA, López-Guerrero JA, Aguilar-Quesada R (2016) DNA source selection for downstream applications based on DNA quality indicators analysis. Biopreserv Biobanking 14:264–270
Mohammadi P, Daneshmand MA, Mahdieh N, Ashrafi MR, Heidari M, Garshasbi M (2021a) Identification of a novel missense c. 386G> A variant in a boy with the POMGNT1-related muscular dystrophy-dystroglycanopathy. Acta Neurol Belg 121:143–151
Mohammadi P, Heidari M, Ashrafi MR, Mahdieh N, Garshasbi M (2021b) A novel homozygous missense variant in the NAXE gene in an Iranian family with progressive encephalopathy with brain edema and leukoencephalopathy. Acta Neurol Belg 121:1–10
Mohammadi P, Salehi Siavashani E, Mohammadi MF, Bahramy A, Almadani N, Garshasbi M (2021c) Whole-exome sequencing identified first homozygous frameshift variant in the COLEC10 gene in an Iranian patient causing 3MC syndrome type 3. Mol Genet Genomic Med 9:e1834
Nerakh G, Ranganath P, Murugan S (2021) Next-generation sequencing in a cohort of Asian Indian patients with the Duchenne muscular dystrophy phenotype: diagnostic yield and mutation spectrum. Thieme 10:023–028
Patel RK, Jain M (2012) NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619
Ren S, Bertels K, Al-Ars Z (2018) Efficient acceleration of the pair-HMMS forward algorithm for GATK HaplotypeCaller on graphics processing units. Evol Bioinforma 14:1176934318760543
Research U (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506–D515
Ryder S, Leadley R, Armstrong N, Westwood M, De Kock S, Butt T, Jain M, Kleijnen J (2017) The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis 12:1–21
Schwartz M, Dunø M (2004) Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Mary Ann Liebert Inc 8:361–367
Sheikh O, Yokota T (2021) Developing DMD therapeutics: a review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies. Expert Opin Investig Drugs 30:167–176
Sheikh O, Yokota T (2020) Advances in genetic characterization and genotype–phenotype correlation of Duchenne and Becker muscular dystrophy in the personalized medicine era. MDPI 10:111
Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DA (2020) The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet 139:1197–1207
Suguna S, Nandal D, Kamble S, Bharatha A, Kunkulol R (2014) Genomic DNA isolation from human whole blood samples by non enzymatic salting out method. Innovare Acad Sci 6:198–199
Takeshima Y, Yagi M, Okizuka Y, Awano H, Zhang Z, Yamauchi Y, Nishio H, Matsuo M (2010) Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet 55:379–388
Tavasoli AR, Memar EHE, Ashrafi MR, Hosseini SMM, Haghighi R, Ghabeli H, Pourbakhtyaran E, Rasoulinezhad M, Mohammadi P, Heidari M (2022) Primary and secondary microcephaly, global developmental delay, and seizure in two siblings caused by a novel missense variant in the ZNF335 gene. J Mol Neurosci 72:1–11
Torella A, Trimarco A, Blanco FDV, Cuomo A, Aurino S, Piluso G, Minetti C, Politano L, Nigro V (2010) One hundred twenty-one dystrophin point mutations detected from stored DNA samples by combinatorial denaturing high-performance liquid chromatography. J Mol Diagn 12:65–73
Verhaart IE, Aartsma-Rus A (2019) Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol 15:373–386
Zamani G, Heidari M, Malamiri RA, Ashrafi MR, Mohammadi M, Badv RS, Hosseini SA, Salehi S, Shahrokhi A, Qorbani M, fathi MR, (2016) The quality of life in boys with Duchenne muscular dystrophy. Neuromuscul Disord 26:423–427
Zamani G, Hosseini Bereshneh A, Azizi Malamiri R, Bagheri S, Moradi K, Ashrafi MR, Tavasoli AR, Mohammadi M, Badv RS, Ghahvechi Akbari M, Heidari M (2020) The first comprehensive cohort of the Duchenne muscular dystrophy in Iranian population: mutation spectrum of 314 patients and identifying two novel nonsense mutations. J Mol Neurosci 70:1565–1573
Acknowledgements
The authors would like to thank the patients and their parents for their cooperation. We also would like to thank all the Iranian Muscular Dystrophy Association members for their assistance in the process of data collection.
Funding
This research is supported by a grant from the Tehran University of Medical Sciences (TUMS). The funding organization had no role in the design and conduct of the study; in the collection, analysis, and interpretation of the data; or in the preparation, review, or approval of the article and the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
GRZ and MFM conceived and designed the experiments. MFM, PM, MH, ART, RH, EPB, HGH, and MRA conducted the experiments. PM, GHZ, MH, and MFM analyzed and interpreted the data. PM and MFM contributed reagents/materials/analysis tools. MFM, PM, MH, SH, and SMH wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
All the patients presented to the Children’s Medical Centre, Tehran University of Medical Sciences (TUMS) or were registered on the Iranian Muscular Dystrophy Association registry system. Written consent was obtained from all the enrolled patients or their respective legal guardians. The research was approved by the local medical ethics committee of TUMS (IRB code: IR.TUMS.MEDICINE.REC.1396.3568). Adherence to the World Medical Association Declaration of Helsinki and its successive revisions was considered in this research.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zamani, G.R., Mohammadi, M.F., Tavasoli, A.R. et al. Genetic Analysis of Forty MLPA-Negative Duchenne Muscular Dystrophy Patients by Whole-Exome Sequencing. J Mol Neurosci 72, 1098–1107 (2022). https://doi.org/10.1007/s12031-022-01980-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-022-01980-5