
Abstract
Delirium is the most common mental disturbance in critically-ill patients and results in significant morbidity and mortality. Haloperidol is a preferred agent for the treatment of delirium in this population because of its rapid onset of action and lack of hemodynamic effects. Despite its widespread use in the critical care setting, most of the relevant data are obtained from case series or extrapolated from non-critically-ill populations. This review provides an overview of haloperidol pharmacokinetics and a comprehensive summary of the evidence for various haloperidol dosing regimens in the treatment of delirium in critically-ill patients. A comprehensive literature search was conducted in Medline, Embase, and International Pharmaceutical Abstracts with “haloperidol”, “delirium”, “agitation”, “critically-ill”, and “intensive care” as keywords. Studies involving haloperidol for delirium prophylaxis, non-critical care settings, and terminally-ill subjects were excluded. Eleven studies were identified: four with intermittent IV haloperidol, four with continuous IV infusion haloperidol, two with oral/enteral haloperidol, and one with IM haloperidol. All of the case reports, case series, and descriptive studies have shown a benefit with haloperidol, but publication bias is likely present. Only three studies were controlled studies, but all had small sample sizes and methodological flaws. Randomized, double-blind, active-comparator trials of haloperidol with allocation concealment are needed. Subsequent research should focus on using validated delirium screening and assessment scales for more objective identification and measurement of delirium outcomes.
Similar content being viewed by others

Introduction
Delirium is defined as change or fluctuation in consciousness characterized by acute onset of impaired cognitive function or perceptual disturbances, and attributed to a general medical condition [1]. Delirium is the most common mental disturbance in the critically-ill and may develop in up to 87% of patients [2, 3]. Although delirium can increase length of hospital stay and health care costs and is an independent predictor for mortality [4–7], its pathogenesis is poorly understood. Causes of delirium are multi-factorial, including: central cholinergic deficiency, cholinergic-dopaminergic imbalance, dopamine excess, inflammation, and/or chronic stress [8–12].
The main goal of therapy is to reverse delirium rapidly without causing adverse effects [13]. Management should always begin with reversing the cause and instituting non-pharmacological measures. If these are insufficient, then pharmacologic treatment is warranted. Haloperidol is a preferred pharmacotherapeutic agent for treating delirium because it targets the pathogenesis of delirium, has a rapid onset of action, no active metabolites, does not require dosage adjustment in organ dysfunction, and has minimal sedative, hypotensive, and autonomic effects [14–19]. Haloperidol can be given orally/enterally (tablet, liquid), intramuscularly, and intravenously. Due to these numerous advantages, haloperidol has been consistently recommended as the agent of choice for delirium in the critically-ill [19]. However, because of very strong binding to D2 receptors in brain dopaminergic pathways, haloperidol can commonly cause akathisia and extrapyramidal symptoms (EPS); it can also cause QTc prolongation [20, 21].
Despite being a first-line agent for delirium, considerable controversy surrounds the use of haloperidol for this indication because of lack of prospective, randomized, placebo-controlled trials [22]. Clinicians have had extensive experience with haloperidol and feel comfortable using it, although data for critically-ill patients are obtained from uncontrolled studies or extrapolated from non-critically-ill populations [15]. Furthermore, intravenous (IV) haloperidol does not have an official indication in Canada or the United States (US) for treating delirium. Recently, the US Food and Drug Administration also advised that IV haloperidol results in increased risk of QTc prolongation, torsades de pointes and death—especially in patients with cardiac or electrolyte abnormalities and on concomitant QTC-prolonging drugs [23].
This narrative review provides an overview of haloperidol pharmacokinetics and summarizes the evidence in terms of efficacy and adverse effect profile of various haloperidol dosing regimens for treating delirium in critically-ill patients.
Methods
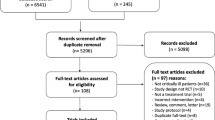
To identify relevant trials evaluating clinical outcomes of various haloperidol dosing strategies for treating critical care delirium, a comprehensive literature search was conducted within the databases of: Medline (1950 to August 2011), Embase (1980 to August 2011), and International Pharmaceutical Abstracts (1970 to August 2011). Keywords included: “haloperidol”, “delirium”, “agitation”, “critically-ill”, and “intensive care”. No search limits were applied and reference lists of relevant articles were reviewed manually. Haloperidol studies concerning delirium prophylaxis, subjects admitted to non-critical care units, and terminally-ill patients were excluded.
Haloperidol Pharmacokinetics and Effects of Physiologic Changes in Critical Illness
Haloperidol has been available in the US since 1967, but the first comprehensive pharmacokinetic study was not published until 1976; [24] few additional studies have been published since [25–31]. Table 1 provides a summary of haloperidol pharmacokinetic parameters [24–31]. Haloperidol pharmacokinetics are best described by a bi- or tri-exponential model. Model-dependent or independent analysis resulted in consistent and comparable estimates for clearance, volume of distribution (V d), and half-life [26]. Following IV or PO administration, distribution half-life (t 1/2α) is 12–60 min with an elimination half-life (t 1/2elim) of 14–20 h [24–29, 31]. Similarly, after IV administration, a rapid first distribution phase (t 1/2α 11–14 min) is followed by slower second distribution phase (t 1/2β 2–4.5 h) and a longer elimination phase (t 1/2elim 26–56 h) [26, 31].
In initial studies involving healthy volunteers or acutely-psychotic patients [24, 26–28], haloperidol demonstrated large inter-individual variability (e.g., coefficients of variation >40%) but minimal intra-individual variability [29, 32]. However, in the critically-ill, it is likely that haloperidol will also exhibit large intra-individual variability because of a fluctuating health status, frequent physiologic alterations, presence of multiple therapeutic interventions, and drug interactions. To our knowledge, haloperidol pharmacokinetics has not been studied in the critically-ill population. We can, however, theorize about the expected changes to conventional pharmacokinetic parameters of haloperidol given our knowledge of physiologic changes in critical illness (see Table 2 for details) [28, 33–42].
Absorption
After intramuscular (IM) and PO administration, time to haloperidol peak plasma concentrations (T max) is 20 min and 1.5–6 h, respectively [24–26, 29, 31]. Absorption half-life (t 1/2abs) of PO administration is 0.25–2 h [24, 26, 28, 31], with an oral bioavailability (F) of 60–70% in the majority of studies [24, 26–28, 31]. Lag time after oral dosing of 0.82–1.3 h has been noted in studies of healthy volunteers and psychotic patients [26, 28].
Rate and extent of absorption are dependent on both chemical properties of haloperidol and milieu at the site of administration (IM and SC) and absorption (PO) [33]. As haloperidol’s chemical properties are constant, the main factor influencing drug absorption in critical illness is physiology at site of absorption (e.g., pH, blood flow, surface area, and gastrointestinal (GI) motility) [33]. Gut and dermal blood flow are reduced because of shunting of blood to vital organs (heart, lungs, brain) during shock states [34]. Prolonged GI dysfunction may also result in intestinal atrophy and decreased surface area for absorption in as little as 3 days [35]. These numerous physiologic changes in critical illness make haloperidol absorption via routes other than IV highly variable. As it is impossible to quantify changes in absorption clinically, IV administration is preferred for most critically-ill patients.
Distribution
Haloperidol is lipophilic and highly protein bound (92%) [24, 43], with extensive tissue distribution and a large V d of 8–22l/kg [26, 28, 31]. Distribution half-life ranges from 12 min to 4.5 h [24, 26, 28, 31]. Elderly or obese patients may have a higher V d because of a higher percentage of body fat. Because of its lipophilicity, haloperidol freely crosses the blood brain barrier. Cerebrospinal fluid concentration can be ten times greater than serum concentrations after IM administration [44].
Rate and extent of drug distribution are dependent on factors such as blood flow, protein binding, tissue permeability, lipophilicity, and degree of ionization [33]. In critical illness, haloperidol distribution to the brain may be affected by changes in blood flow and protein binding. Shock states result in global hypoperfusion, but haloperidol delivery to brain may be unchanged because of the shunting of blood to vital organs during shock. However, in severe shock states, even this relative increase is less than normal physiologic blood flow and can result in suboptimal haloperidol delivery. Once haloperidol reaches its target, passage into tissue simply requires passive diffusion of free (unbound) drug [43].
Metabolism
With a clearance of 8–15 ml/min/kg [24, 26, 28, 31], haloperidol undergoes predominantly hepatic metabolism via the cytochrome P450 system. As substrate and inhibitor of CYP 3A4 and substrate of CYP 2D6, haloperidol undergoes Phase I metabolism reactions of oxidative dealkylation to form inactive metabolites and reduction to form an active metabolite (reduced haloperidol) [24, 32]. As expected from first-pass metabolism, oral (compared to IV) administration is associated with greater formation of reduced haloperidol [31, 45]. Reduced haloperidol exhibits only 1/400 of parent drug pharmacological activity [46]; whether this activity is exerted by the reduced form itself or is because of re-conversion to parent compound in vivo is unclear [32, 45, 47]. Haloperidol has an intermediate extraction ratio (E 0.3–0.7) [26, 28]. As haloperidol metabolism primarily occurs in liver, total body clearance of haloperidol is approximated by hepatic clearance and changes in hepatic blood flow, intrinsic clearance, or protein binding can impact haloperidol clearance.
Excretion
As haloperidol almost exclusively undergoes hepatic metabolism, only 1% is excreted unchanged in urine [32]. Changes in renal physiology and decreased renal blood flow in critically-ill patients have minimal effect on haloperidol pharmacokinetic parameters overall. Hence, haloperidol may be administered safely in patients with renal dysfunction without dosage adjustment.
Summary of Pharmacokinetics
Many physiologic changes occur in the critically-ill patient which may affect haloperidol pharmacokinetics. Hypoperfusion from shock accounts for most ADME changes in critical illness as it decreases absorption of oral haloperidol and distribution to target and metabolizing organs. However, given that haloperidol’s pharmacologic effect is best assessed using standardized sedation and delirium scales and not plasma haloperidol concentrations, these changes may have little clinical relevance.
Results of Literature Search for Evidence on Various Haloperidol Dosing Regimens
Eleven studies of different haloperidol dosing strategies for delirium treatment in critically-ill patients were identified: four with intermittent IV infusion [48–51], four with continuous IV infusion [18, 52–54], two with oral/enteral [55, 56], and one with IM [57]. Tables 3 [18, 48–53, 57], and 4 [54–56] summarize the descriptive and comparative studies, respectively.
Intermittent IV Administration
The IV route of administration may be preferred when the oral route is inaccessible because of the presence of endotracheal intubation or if patients have a non-functional GI tract. It may also be favorable since it takes a considerable amount of time to prepare and administer medications enterally. In intensive care units (ICUs), intermittent IV infusion remains the most commonly-used administration regimen of haloperidol because of its predicable pharmacokinetic profile compared to other routes, ease of administration, fast onset of action, and minimal sedation and hypotension. Historically, two approaches to intermittent IV dosing include plateau dosing schemes and escalating dosage regimens [58]. Plateau dosing involves administering the same repeated dose if there is inadequate response to the previous dose, whereas escalating dosage involves doubling the dose every 15–20 min until therapeutic effect is reached. Some references state that individual doses <50 mg are sufficient, while others believe that doses >10 mg/h offer no additional benefit [8]. Four relevant studies of intermittent IV dosing of high-dose haloperidol to treat delirium in critically-ill patients were identified. One is a case series [48], two are descriptive studies [49, 50], and one is a case report [51].
In the case series, Tesar et al. [48]. described four cases (3 male) of high-dose intermittent IV haloperidol use in cardiac care unit (CCU) patients from July 1983 to June 1984. Patients were 50–62 years-old with life-threatening coronary artery disease warranting intra-arterial balloon pump (IABP) insertion. All patients exhibited delirium and agitation symptoms within a few days after admission that did not resolve with benzodiazepines or low-dose haloperidol (5–10 mg). All required escalating bolus doses (30–75 mg) every 15–60 min to control delirium, with maximum dosages ranging from 140 mg/day to 485 mg over 8 h. Once delirium was controlled, dosing interval was increased to every 3–4 h. Delirium resolved in all patients once they no longer required IABP for hemodynamic support; haloperidol and sedation were discontinued shortly post-IABP removal. No patients exhibited EPS or adverse cardiac effects during their hospital stay.
Adams et al. [49] described their experiences with high-dose haloperidol and lorazepam combination to treat delirium in medical and surgical ICU patients. The authors described 25 advanced cancer patients (10 male) with median age of 54 (range 22–75 years) and delirium based on DSM-III criteria; all had at least two serious medical complications (e.g., respiratory failure, sepsis, myocardial infarction, hepatic or renal toxicity). Sixteen patients were intubated, nine of whom were receiving dopamine for hypotension and five had recently undergone major surgery (i.e., abdominal, thoracic, head, and neck). Total 24-h dose of IV haloperidol and lorazepam ranged from 100–480 to 36–480 mg, respectively. Maximal dose of each medication was 10 mg every hour, consecutively for 15 days, with the longest treatment interval being 3 months. In most patients (24/25), sedation was achieved within 90 min. Eighteen patients recovered from delirium: six after haloperidol and lorazepam administration and twelve after reversal of underlying causes. The other seven passed away in ICU within 2 days to 2 months after developing delirium; in these patients, haloperidol and lorazepam doses were generally higher and used as palliative sedation. No patients experienced respiratory, cardiac, or hemodynamic complications, but one experienced dystonia during weaning from high- to low-dose haloperidol.
Another descriptive study of delirium treatment with IV haloperidol in surgical ICU patients was conducted by Moulaert [50]. Patients whose delirium may have been attributed to alcohol use, pain, or sepsis were excluded. Six post-operative vascular surgery patients (5 male), with mean age of 74 ± 5 years were included and on dosing protocols, where haloperidol was started at 5 mg and doubled every 30 min until the patient was calm; if the patient remained calm for >30 min but agitation reappeared, the protocol was repeated again starting with 5 mg. At baseline, all patients were agitated, but all had resolution of their symptoms after IV haloperidol treatment. Each patient required an average of two protocol cycles to control their delirium, with a mean dose of 19 mg per cycle. Agitation usually improved within 60 min. No side effects were noted in any patient.
Sanders et al. [51] described a case of a 56 year-old male with a history of cardiac disease admitted to the CCU for unstable, refractory angina. Within 3 h of IABP placement, he became agitated, disoriented, and displayed verbal and physical aggression that only resolved with IV haloperidol 50 mg hourly; this dose was continued hourly in addition to lorazepam 2 mg every 2–4 h for the next 48 h. The patient received haloperidol 1,200 and 1,100 mg in the first and second 24 h periods, respectively. Post-IABP removal on day 6, IV haloperidol 320 and 450 mg were administered daily for the next 2 days before extubation. The patient exhibited no signs of akathisia or EPS, although his QTc interval varied from 400 to 584 ms without relationship to haloperidol dosing.
In summary, high-dose intermittent IV haloperidol dosing at frequent intervals is effective in controlling delirium in critically-ill patients, especially for those refractory to other treatment modalities. Most dosing schemes were escalating dosage regimens. However, these studies consisted of case reports and descriptive studies with no comparative studies. In addition, studies are dated (with publication dates ranging from 1985 to 1991) and authors were not explicit on methods of assessing delirium nor frequency of assessment [48–51]. Studies also focused mainly on agitated delirium, and in most cases, it was difficult to determine whether the patient was agitated or acutely-delirious. Regarding adverse events, EPS or cardiac rhythm abnormalities were infrequently noted, but IV haloperidol administration is speculated to result in less EPS than oral administration because of lack of first-pass metabolism [59]. In addition, most patients received concomitant benzodiazepines, which could worsen or prolong time in delirium and mask EPS. It is unclear as to whether EPS were actively monitored, which is important as it would be easy for EPS to go unrecognized in mechanically-ventilated critically-ill patients.
Continuous IV Infusion
Considering its long terminal half-life, it may not seem intuitive to use haloperidol as a continuous infusion. However, the literature generally indicates that patients with delirium in ICU settings tend to require larger haloperidol doses compared to non-critically-ill patients [13]. In the studies investigating high-dose intermittent IV infusion [48–51], some patients required haloperidol boluses as frequently as every 30 min. The time it takes to resolve delirium and prepare and administer boluses at this frequency can be extremely labor intensive for nursing and paramedical staff. Other proposed advantages of haloperidol continuous infusion over intermittent IV administration include: less cyclical agitation and sedation, less over-sedation, and potential facilitation of ventilator weaning [18]. Four studies examined haloperidol continuous infusion to control delirium in critically-ill patients [18, 52–54]. One is a case report [52], one a retrospective, cross-sectional chart review [18], one a case series [53], and one a randomized, open-label, parallel group study [54].
Fernandez et al. [52] described the case of a previously healthy 45 year-old female admitted for vaginal bleeding from untreated cervical carcinoma. She became mildly anxious and restless on day 14, which progressed to combativeness the next day despite low doses of haloperidol and lorazepam; no reversible etiology of delirium could be found. On day 16, she was treated with increasing doses of intermittent IV haloperidol (10–20 mg/h), lorazepam (4–8 mg/h), and hydromorphone (2–4 mg/h) without effect; the longest period of calmness the patient experienced ranged only 10–20 min. Increasing the haloperidol dose to 25 mg/h (600 mg/day) finally controlled the patient adequately to allow for discontinuation of lorazepam. The haloperidol infusion was discontinued after 5 days and replaced by bedtime doses of IV haloperidol (20 mg) and lorazepam (4 mg) without agitation recurrence.
The largest cohort of patients receiving haloperidol continuous infusion is described by Riker et al. [18]. in a retrospective, cross-sectional chart review of all patients who required continuous infusion of haloperidol from January to June 1992. At the time of study, sedation scales that spanned a small number of severity categories or had a symmetrical range of severity for both sedation and agitation were not yet developed. Thus, the authors developed the Sedation Agitation Scale (SAS), where −3 = unarousable, −2 = very oversedated, −1 = oversedated, 0 = calm and cooperative, +1 = agitated, +2 = dangerously agitated, and +3 = immediate threat to safety; a score of −1 to +1 was considered acceptable. Eight patients (4 male) were identified, with mean age of 47 years, mean APACHE II score of 24, and mean ICU length of stay of 25 days. Admission diagnoses included cardiogenic shock, respiratory failure, and drug overdose. Haloperidol infusions were started after a mean 2.0 ± 1.7 days of intermittent IV infusion, and on ICU day 15 ± 13.6. Infusion rates ranged from 3 to 40 mg/h with mean starting rate of 9 ± 7 mg/h and maximum rate of 18 ± 11 mg/h for mean duration of 7 ± 3.2 days (range 3–12 days). Maximum haloperidol daily dose during continuous infusion was 398 ± 248 mg (range 75–865 mg). After 1 day of continuous infusion, mean daily haloperidol dose increased from 68 ± 59 to 269 ± 178 mg, whereas sedative requirements and daily supplemental sedative doses also decreased. The SAS scores also decreased from baseline mean score of +2.4 to +1.8 and +0.8 after 1 and 2 days of continuous infusion, respectively. Three patients experienced four complications potentially related to continuous haloperidol infusion, including: atrial ectopic activity and intermittent third degree atrioventricular block; prolonged QTc (0.45–0.64 s) that resolved upon haloperidol discontinuation; monomorphic ventricular tachycardia; and tremors that developed after lorazepam was discontinued.
Seneff and Matthews [53] described three critically-ill patients (2 male), 32–66 years-old who were admitted to ICU for trauma-related injuries or respiratory failure and required continuous infusion of haloperidol to control delirium symptoms; two patients had history of schizophrenia. All patients exhibited extreme agitation and unresolved delirium despite non-pharmacological interventions, concomitant IV infusions of benzodiazepines and opioids, and IV boluses of haloperidol; two patients also required neuromuscular blocking agents. Haloperidol 10 mg boluses were initiated and later increased to 4–24 mg/h infusions. Continuous haloperidol infusions resulted in rapid control of delirium and agitation, and allowed for sedative discontinuation and ventilator weaning. No major adverse effects were noted with infusions, except in one patient where three doses of diphenhydramine 25 mg were administered for possible akathisia symptoms.
In the most recent trial of haloperidol continuous infusion (a randomized, open-label, parallel group pilot study), Reade et al. [54] compared dexmedetomidine versus haloperidol for treating delirium in 20 intubated patients. Patients were eligible if agitation was the only barrier to extubation. Eligible patients were randomized to dexmedetomidine 0.2–0.7 mcg/kg/h infusion with or without a 1.0 mcg/kg loading dose at physician’s discretion, or haloperidol 0.5–2 mg/h with or without a 2.5 mg loading dose. Dose increases were determined by monitoring the Richmond Agitation and Sedation Scale (RASS) every 4 h, with a goal score of 0. Primary endpoint was time from start of study drug to extubation. Twenty patients aged 42–78 years were enrolled with mean APACHE II score of 14, and admission diagnoses of pneumonia, sepsis, post-cardiothoracic surgery, and post-neurosurgery. At enrollment, 30–40% had an Intensive Care Delirium Screening Checklist (ICDSC) score of ≥4 (i.e., positive screen for delirium). Patients on dexmedetomidine were extubated sooner than patients on haloperidol (19.9 h vs. 42.2 h, P = 0.016). However, more patients continued to receive dexmedetomidine compared to haloperidol post-extubation (7 vs. 4 patients). Patients receiving dexmedetomidine were also discharged from the ICU sooner (1.5 d vs. 5.5 d, P = 0.0039) and achieved goal RASS scores more quickly, but there were no differences in supplemental sedation requirements between the groups. QTc was prolonged in patients on haloperidol versus dexmedetomidine (0.446 s vs. 0.395 s, P = 0.0061) although this difference may not be clinically relevant.
In summary, continuous infusions of haloperidol appear to control delirium and agitation in patients with an inadequate response to intermittent IV infusions. However, patients from descriptive, non-comparative studies may not have responded to intermittent IV administration of haloperidol because they were also receiving high-dose opioids and benzodiazepines [18, 52, 53]. Continuous infusions offer advantages of ease of administration and potentially decreased nursing labor and costs. Riker et al. [18]. calculated that beginning continuous infusion resulted in median daily nursing time savings of 154 min. However, when prospectively compared to dexmedetomidine, haloperidol continuous infusion appeared less efficacious for treating delirium [54]. Although strengths of this trial include use of a comparator group and validated sedation and delirium scales, only hyperactive delirium was studied. Other limitations include small sample size, lack of ventilator weaning protocols, and no mention of non-pharmacological approaches to delirium. Continuous infusion of haloperidol does not offer pharmacokinetic advantages either since haloperidol already has a long elimination half-life. Also, continuous infusions would be expected to result in higher incidence of adverse effects because of higher daily dose and prolonged drug accumulation. Increased adverse events were not noted in the studies summarized [18, 52–54] but assessments were not performed consistently. This is important as adverse effects (e.g., EPS) may not be identified in the critically-ill without regular monitoring and akathisia may be misdiagnosed as worsening agitation.
Oral/Enteral Route
Oral dosage forms may be a reasonable option if the patient has a functional GI tract; but unpredictable absorption and potential for more adverse effects (i.e., EPS) still limit the use of this dosage form in the critically-ill population [59]. Oral haloperidol has longer T max compared to IV haloperidol (2–3 h vs. 20 min). Given the goal to rapidly reverse delirium, administering haloperidol orally could delay delirium reversal in acutely-delirious patients, although some may argue that the oral/enteral route offers a longer duration of action compared to IV. Two studies using the oral route for haloperidol were identified [55, 56]. One compared oral haloperidol to oral risperidone [55], and the other compared oral/enteral haloperidol to oral/enteral olanzapine [56].
To compare the clinical efficacy of risperidone versus haloperidol for delirium treatment, Han and Kim [55] conducted a double-blind, randomized study of 24 delirious patients from medical, intensive care, and oncology units at one Korean institution. Eligible patients had positive delirium screens based on Confusion Assessment Method (CAM) and Delirium Rating Scales (DRS) and had to meet diagnostic criteria for delirium according to DSM-III-R. Patients initially received oral risperidone 0.5 mg or haloperidol 0.75 mg twice daily. Dosages were increased according to Memorial Delirium Assessment Scale (MDAS) results conducted daily at the same time by a blinded psychiatrist. Response was defined as MDAS score <13. Initial DRS scores for all subjects were 22.76 ± 4.30; a score of ≥19 is considered diagnostic for delirium. Outcomes of interest were not explicitly stated, but at study conclusion (7 days), there were no statistically significant differences in any comparisons between groups including mean MDAS scores (P = 0.51), change in DRS scores (risperidone 23.50 vs. haloperidol 21.83, P = 0.35), frequency of response to drugs (risperidone 42% vs. haloperidol 75%, P = 0.11), or average time to response (risperidone 4.17 d vs. haloperidol 4.22, P = 0.95). Mean daily doses of risperidone and haloperidol were 1.02 ± 0.41 mg and 1.71 ± 0.84 mg, respectively. No study participants experienced clinically significant side effects, except one subject on haloperidol had akathisia that was tolerable for the study duration. However, the study duration was short and mean daily haloperidol doses were relatively low.
To compare the safety and response of oral/enteral olanzapine versus oral/enteral haloperidol in treatment of ICU delirium, Skrobik et al.[56] conducted a prospective randomized trial involving 103 ICU patients, of whom 83 were included in the final analysis. Patients were eligible if they received a score of ≥4 on the ICDSC or had clinical manifestations of delirium; diagnosis was confirmed using DSM-IV criteria. Patients, predominantly surgical, were on average 63–68 years-old with mean APACHE II scores of 12–14. Patients were randomized to olanzapine 5 mg daily or haloperidol 2.5–5 mg every 8 h with doses titrated to the Ramsay sedation scale. Intermittent IV haloperidol as rescue medication could be administered to any patient who developed agitation during the study period. There was no explicit primary outcome, but the following variables were measured at baseline and up to 5 days: daily dose of rescue haloperidol, sedative use, and anti-Parkinson medications for EPS; Delirium Index (DI); and daily worst Ramsay score. EPS were assessed with Ross-Chouinard and Angus-Simpson scales. At study conclusion, mean daily doses of olanzapine and haloperidol were 4.54 mg (range 2.5–13.5 mg) and 6.5 mg (range 1–28 mg), respectively. Proportion of patients requiring rescue haloperidol, dose of rescue haloperidol in each group, reduction in DI scores and Ramsay scores were similar between groups. On average, DI decreased from 7.08 to 5.05 from day 1 to 5. No patients received anti-Parkinson medications, but 6 in the haloperidol group exhibited mild EPS compared to none in the olanzapine group.
In summary, studies comparing low doses of oral haloperidol with oral risperidone or olanzapine did not show any differences in delirium treatment [55, 56]. Han and Kim study [55] was in a diverse hospital population, but the percentage of critically-ill patients enrolled in the trial was not mentioned. Allocation concealment is unknown, and also uncertain is whether the study was powered to detect differences in MDAS scores, although the change in MDAS scores was similar in both groups. Skrobik et al. study [56] is the only trial of haloperidol in critically-ill patients that used validated scales for both delirium and EPS assessment. However, allocation concealment was not performed as randomization was conducted on an even/odd day basis. In addition, haloperidol doses used in these trials were lower than doses in case reports and descriptive studies of intermittent IV and continuous infusion. More EPS was noted in the haloperidol group, especially when EPS was actively monitored using objective scales [56].
Intramuscular Route
Intravenous administration may be difficult in severely delirious and combative patients; thus, IM administration of haloperidol is a logical alternative. Intramuscular administration may also be preferred in settings that lack routine cardiac monitoring (e.g., emergency departments). In a pharmacokinetic study conducted by Schaffer et al. with schizophrenic patients, IM administration of haloperidol resulted in faster T max and higher AUC than oral administration [25, 32].
Only one study with IM haloperidol in critically-ill patients was identified [57]. Moore described the case of a 34 year-old male admitted to ICU and mechanically-ventilated because of multiple traumatic injuries post-motor vehicle accident. On day 8, he became agitated and physically combative. Reversible causes of delirium were ruled out before he received IM haloperidol 10 mg hourly. The patient achieved remission after 30 mg of IM haloperidol without adverse effects (i.e., hypotension, EPS). An enteral dose of haloperidol (15 mg twice daily) was then started along with IM haloperidol 5 mg IM hourly as needed. Although, the patient required frequent supplementary doses of IM haloperidol, these gradually decreased to zero on day 14. Haloperidol was discontinued on day 19 and delirium did not recur.
This case report suggests that IM administration of haloperidol was effective for controlling delirium with no adverse effects in a critically-ill patient; this is similar to findings in non critically-ill acutely-psychotic patients [60–62]. However, it may be unnecessary to administer IM haloperidol to critically-ill patients given that they often have established IV access. Given limited data, IM administration should be considered only when IV administration is unavailable or contraindicated.
Discussion
Although haloperidol has been consistently recommended in guidelines as first-line treatment for delirium in critically-ill patients [19], minimal robust evidence exists in the literature to support its efficacy [21]. If the different dosage strategies of haloperidol are categorized based on quality of evidence, then oral haloperidol would have the highest level (with two active-comparator trials), followed by continuous infusion (one active-comparator trial), then intermittent infusion (multiple case reports and descriptive studies) and lastly, IM administration (one case report). All case reports, case series, and descriptive studies have reported a benefit with haloperidol, but publication bias may be present. Oral haloperidol was not significantly different when compared to olanzapine or risperidone, and continuous infusion of haloperidol was less efficacious when compared to dexmedetomidine in a pilot study. However, small sample size and methodological flaws limit these comparative studies.
Milbrandt et al. conducted a retrospective cohort study of mechanically-ventilated patients who received haloperidol within 2 days of mechanical ventilation (N = 83) compared to those who had not received haloperidol during their hospital stay (N = 906) [63]. The authors found decreased mortality in the haloperidol group (20.5% vs. 36.1%, P = 0.004), and this result persisted after adjusting for age, comorbidities, severity of illness, organ dysfunction, admitting diagnosis and other confounders (OR 0.35; 95% confidence interval 0.18–0.69; P-value 0.0022). As delirium was not an inclusion criteria, the reduction in mortality in the haloperidol group could have been a function of treating delirium—a condition associated with increased mortality in the critically-ill (confounding by indication). We were unable to include this study in our review as the routes of administration of haloperidol were not specified. However, the results of this study are hypothesis generating and notable as it was the first study to demonstrate decreased mortality with haloperidol use. There is great need for more robust research in the area of delirium, as a recent Cochrane systematic review identified only three un-confounded, randomized trials of antipsychotics in delirium [16].
Furthermore, all descriptive studies identified have publication dates ranging from 1977 to 1995 [18, 48–53, 57]. Care of critically-ill patients and our understanding and approach to delirium screening and treatment have improved substantially over the past 30 years, limiting application of these study results to the critically-ill patient today. In all but two studies published before 2000, no objective measure of delirium was used and criteria for “response” were not clearly described. Objective measure of delirium is important in the critically-ill, as delirium may present as three distinct subtypes: hyperactive; hypoactive; or mixed hyperactive-hypoactive state. Studies of haloperidol in critically-ill patients have typically focused on hyperactive delirium, but the majority of delirious patients present with the hypoactive subtype. Hypoactive delirium is significantly under diagnosed in practice, as it may be missed 70% of the time if clinicians fail to use a screening tool for delirium [5, 64]. Another reason for an objective screening tool is to differentiate between agitation symptoms and delirium in the ICU setting, particularly in mechanically-ventilated patients [13]. Although agitation and delirium are often used interchangeably, agitation is a symptom with multiple etiologies, including delirium, pain, anxiety, metabolic abnormalities, withdrawal, and drug toxicity.
Delirium is also a recent term used in the critically-ill, as patients presenting with similar symptom in the past were referred to as having “ICU psychosis”. Other terms that have been used interchangeably include anxiety and agitation. Lack of standardization in terminology for delirium makes literature interpretation difficult, as appropriate treatment primarily depends on accurate diagnosis derived from validated screening and assessment tools. In addition, no study appeared to implement non-pharmacologic therapies as adjunct to pharmacologic treatment. Pharmacotherapy is considered as only one facet of delirium treatment, as non-pharmacologic measures such as re-orientation, physiotherapy, and sleep hygiene are considered equally important.
Interestingly, despite large doses of haloperidol used in the studies with critically-ill patients, few adverse effects were noted. The high haloperidol doses in these studies may be attributed to concomitant administration of large doses of opioids and benzodiazepines, which worsen delirium. Potential reasons for lack of adverse effects include: lack of active monitoring; no first-pass metabolism with IV and IM routes; and concomitant benzodiazepine use. Only one study used objective scales to detect EPS [56]. In non-critically-ill populations, EPS are typically identified by the patient; however, it is challenging to rely on mechanically-ventilated, critically-ill patients to report symptoms. In addition, for patients receiving intermittent neuromuscular blockade, EPS cannot be objectively diagnosed. The highest incidence of EPS were found in the study that utilized a validated rating scale [56]. Non-oral routes of haloperidol administration are associated with lower EPS rates than oral routes; the mechanism of this observation is unknown but is speculated to be because of the lack of first-pass metabolism [59]. Lastly, most critically-ill patients receive intermittent or regular benzodiazepines therapy for various indications which may mask the presence of EPS. Regarding QTc prolongation, despite most critically-ill patients being on telemetry, this side effect may not be proactively monitored.
Given known toxicities of haloperidol, new agents (i.e., atypical antipsychotics and dexmedetomidine) that are potentially equally efficacious with fewer side effects represent alternate pharmacotherapeutic options for treating delirium. Atypical antipsychotics, risperidone, and olanzapine, have shown equal efficacy with similar incidence of adverse effects as compared to low-dose haloperidol [55, 56]. Given the growing body of evidence supporting their use, atypical antipsychotics may be considered first-line in treatment of delirium for patients refractory to low-dose haloperidol or who are at risk of adverse effects from haloperidol (i.e., prolonged QT interval or EPS at baseline, cardiac abnormalities, on concomitant QT-prolonging drugs) [16]. However, one limitation of atypical antipsychotics is the lack of non-depot parenteral dosage forms; oral dosage forms may be unfavorable in patients with compromised GI physiology and who require rapid control of acute delirium. Although, IM olanzapine is available commercially, it is not readily accessible in all hospitals.
In summary, IV regimens of haloperidol are most useful in the critically-ill given their fast onset of action, ease of administration, and predictable pharmacokinetics. Intermittent IV administration should be the dosing regimen of choice, whereas continuous infusion of haloperidol may be considered for patients failing to respond to large, frequent boluses of IV haloperidol. Oral/enteral haloperidol may be used as maintenance regimen in patients with a functional GI tract. Intramuscular haloperidol may be considered in the acutely-combative patient without IV access; however, literature in the critically-ill is sparse regarding this administration route. All haloperidol dosage forms have low incidence of adverse effects like EPS or QTc prolongation; however, safety assessments were not done routinely in earlier studies and most patients received concomitant benzodiazepines. It is important for clinicians to proactively and regularly monitor for these side effects when administering any dose or route of haloperidol.
Conclusion
Regardless of dosage strategy of haloperidol used, it is important to investigate the underlying etiology, reversed it if possible, and always use non-pharmacologic strategies in conjunction with pharmacologic therapy in the management of delirium. Prospective, double-blind, active-comparator trials with allocation concealment are urgently needed with haloperidol, and future research should focus on using validated delirium screening and assessment scales for more objective identification and measurement of delirium outcomes.
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders—text revision. Delirium, dementia, and amnestic and other cognitive disorders. 4th ed. Washington: American Psychiatric Association; 2000.
Fish DN. Treatment of delirium in the critically ill patient. Clin Pharm. 1991;10:456–66.
Pisani MA, McNicoll L, Inouye SK. Cognitive impairment in the intensive care unit. Clin Chest Med. 2003;24:727–37.
Milbrandt EB, Deppen S, Harrison PL, et al. Costs associated with delirium in mechanically ventilated patients. Crit Care Med. 2004;32:955–62.
Ely EW, Shintani A, Truman B, et al. Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA. 2004;291:1753–62.
Ely EW, Gautam S, Margolin R, et al. The impact of delirium in the intensive care unit on hospital length of stay. Intensive Care Med. 2001;27:1892–900.
McNicoll L, Pisani MA, Zhang Y, et al. Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51:591–8.
Adams F. Emergency intravenous sedation of the delirious, medically ill patient. J Clin Psychiatry. 1988;49(Suppl):22–7.
Hshieh TT, Fong TG, Marcantonio ER, et al. Cholinergic deficiency hypothesis in delirium: a synthesis of current evidence. J Gerontol A Biol Sci Med Sci. 2008;63:764–72.
Tune L. The role of antipsychotics in treating delirium. Curr Psychiatry Rep. 2002;4:209–12.
Van Eijk M, Slooter A. Delirium in intensive care unit patients. Semin Cardiothorac Vasc Anesth. 2010;14:141–7.
Han L, McCusker J, Cole M, et al. Use of medications with anticholinergic effect predicts clinical severity of delirium symptoms in older medical inpatients. Arch Intern Med. 2001;161:1099–105.
Hassan E, Fontaine DK, Nearman HS. Therapeutic considerations in the management of agitated or delirious critically ill patients. Pharmacotherapy. 1998;18:113–29.
Settle EC, Ayd FJ. Haloperidol: a quarter century of experience. J Clin Psychiatry. 1983;44:440–8.
Lacasse H, Perreault MM, Williamson DR. Systematic review of antipsychotics for the treatment of hospital-associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40:1966–73.
Lonergan E, Britton AM, Luxenberg J, Wyller T. Antipsychotics for delirium. Cochrane Database Syst Rev. 2007;2:CD005594. http://www.cochrane.org/cochrane-reviews/citing-our-products.
Campbell N, Boustani MA, Ayub A, et al. Pharmacological management of delirium in hospitalized adults—a systematic evidence review. J Gen Intern Med. 2009;24:848–53.
Riker RR, Fraser GL, Cox PM. Continuous infusion of haloperidol controls agitation in critically ill patients. Crit Care Med. 1994;22:433–40.
Jacobi J, Fraser GL, Coursin DB, et al. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med. 2002;30:119–41.
e-CPS. Haloperidol CPhA Monograph. Canadian Pharmacists Association. http://www.e-therapeutics.ca/cps.showMonograph.action?simpleMonographId=m244500#m244500n00011 (2009) Accessed 13 June 2011.
Granger B, Albu S. The haloperidol story. Ann Clin Psychiatry. 2005;17:137–40.
Ryan CJ. Optimising management of delirium. Placebo controlled trials of pharmacological treatments are needed. BMJ. 2001;322:1602.
US Food and Drug Administration. Drug Safety Information for Healthcare Professionals—Information for Healthcare Professionals: Haloperidol (marketed as Haldol, Haldol Decanoate and Haldol Lactate). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHealthcareProfessionals/ucm085203.htm (2009). Accessed 13 June 2011.
Forsman A, Ohman R. Pharmacokinetic studies on haloperidol in man. Curr Ther Res Clin Exp. 1976;20:319–36.
Schaffer CB, Shahid A, Javaid JI, et al. Bioavailability of intramuscular versus oral haloperidol in schizophrenic patients. J Clin Psychopharmacol. 1982;2:274–7.
Holley FO, Magliozzi JR, Stanski DR, et al. Haloperidol kinetics after oral and intravenous doses. Clin Pharmacol Ther. 1983;33:477–84.
Magliozzi JR, Hollister LE. Elimination half-life and bioavailability of haloperidol in schizophrenic patients. J Clin Psychiatry. 1985;46:20–1.
Cheng YF, Paalzow LK, Bondesson U, et al. Pharmacokinetics of haloperidol in psychotic patients. Psychopharmacology. 1987;91:410–4.
Midha KK, Chakraborty BS, Ganes DA, et al. Intersubject variation in the pharmacokinetics of haloperidol and reduced haloperidol. J Clin Psychopharmacol. 1989;9:98–104.
Chakraborty BS, Hubbard JW, Hawes EM, et al. Interconversion between haloperidol and reduced haloperidol in healthy volunteers. Eur J Clin Pharmacol. 1989;37:45–8.
Chang WH, Lam YW, Jann MW, et al. Pharmacokinetics of haloperidol and reduced haloperidol in Chinese schizophrenic patients after intravenous and oral administration of haloperidol. Psychopharmacology. 1992;106:517–22.
Froemming JS, Lam YW, Jann MW, et al. Pharmacokinetics of haloperidol. Clin Pharmacokinet. 1989;17:396–423.
Boucher BA, Wood GC, Swanson JM. Pharmacokinetic changes in critical illness. Crit Care Clin. 2006;22:255–71.
De Paepe P, Belpaire FM, Buylaert WA. Pharmacokinetic and pharmacodynamic considerations when treating patients with sepsis and septic shock. Clin Pharmacokinet. 2002;41:1135–51.
Hernandez G, Velasco N, Wainstein C, et al. Gut mucosal atrophy after a short enteral fasting period in critically ill patients. J Crit Care. 1999;14:73–7.
Arana GW, Goff DC, Friedman H, et al. Does carbamazepine-induced reduction of plasma haloperidol levels worsen psychotic symptoms? Am J Psychiatry. 1986;143(5):650–1.
Jann MW, Ereshefsky L, Saklad SR, et al. Effects of carbamazepine on plasma haloperidol levels. J Clin Psychopharmacol. 1985;5:106–9.
Kidron R, Averbuch I, Klein E, et al. Carbamazepine-induced reduction of blood levels of haloperidol in chronic schizophrenia. Biol Psychiatry. 1985;20:219–22.
Linnoila M, Viukari M, Vaisanen K, et al. Effect of anticonvulsants on plasma haloperidol and thioridazine levels. Am J Psychiatry. 1980;137:819–21.
Takeda M, Nishinuma K, Yamashita S, et al. Serum haloperidol levels of schizophrenics receiving treatment for tuberculosis. Clin Neuropharmacol. 1986;9:386–97.
Jann MW, Saklad SR, Ereshefsky L, et al. Effects of smoking on haloperidol and reduced haloperidol plasma concentrations and haloperidol clearance. Psychopharmacology. 1986;90:468–70.
McKindley D, Hanes S, Boucher B. Hepatic drug metabolism in critical illness. Pharmacotherapy. 1998;18:759–78.
Javaid J. Clinical pharmacokinetics of antipsychotics. J Clin Pharmacol. 1994;34:286–95.
Beresford R, Ward A. Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis. Drugs. 1987;33:31–49.
Volavka J, Cooper TB. Review of haloperidol blood level and clinical response: looking through the window. J Clin Psychopharmacol. 1987;7:25–30.
Korpi ER, Wyatt RJ. Reduced haloperidol: effects on striatal dopamine metabolism and conversion to haloperidol in the rat. Psychopharmacology. 1984;83:34–7.
Shostak M, Perel JM, Stiller RL, et al. Plasma haloperidol and clinical response: a role for reduced haloperidol in antipsychotic activity? J Clin Psychopharmacol. 1987;7:394–400.
Tesar GE, Murray GB, Cassem NH. Use of high-dose intravenous haloperidol in the treatment of agitated cardiac patients. J Clin Psychopharmacol. 1985;5:344–7.
Adams F, Fernandez F, Andersson BS. Emergency pharmacotherapy of delirium in the critically ill cancer patient. Psychosomatics. 1986;27(Suppl 1):33–8.
Moulaert P. Treatment of acute nonspecific delirium with i.v. haloperidol in surgical intensive care patients. Acta Anaesthesiol Belg. 1989;40:183–6.
Sanders KM, Murray GB, Cassem NH. High-dose intravenous haloperidol for agitated delirium in a cardiac patient on intra-aortic balloon pump. J Clin Psychopharmacol. 1991;11:146–7.
Fernandez F, Holmes VF, Adams F, et al. Treatment of severe, refractory agitation with a haloperidol drip. J Clin Psychiatry. 1988;49:239–41.
Seneff MG, Mathews RA. Use of haloperidol infusions to control delirium in critically ill adults. The Annals of pharmacotherapy. 1995;29:690–3.
Reade MC, O’Sullivan K, Bates S, et al. Dexmedetomidine versus haloperidol in delirious, agitated, intubated patients: a randomised open-label trial. Crit Care. 2009;13:R75.
Han CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45:297–301.
Skrobik YK, Bergeron N, Dumont M, et al. Olanzapine versus haloperidol: treating delirium in a critical care setting. Intensive Care Med. 2004;30:444–9.
Moore DP. Rapid treatment of delirium in critically ill patients. Am J Psychiatry. 1977;134:1431–2.
Fraser GL, Riker RR. Controlling severe agitation in the critically ill with intravenous haloperidol. Hosp Pharm. 1994;29:689–91.
Menza MA, Murray GB, Holmes VF, et al. Decreased extrapyramidal symptoms with intravenous haloperidol. J Clin Psychiatry. 1987;48:278–80.
Man PL, Chen CH. Rapid tranquilization of acutely psychotic patients with intramuscular haloperidol and chlorpromazine. Psychosomatics. 1973;14:59–63.
Anderson WH, Kuehnle JC, Catanzano DM. Rapid treatment of acute psychosis. Am J Psychiatry. 1976;133:1076–8.
Carter RG. Psychotolysis with haloperidol. Rapid control of the acutely disturbed psychotic patient. Dis Nerv Syst. 1977;38:237–9.
Milbrandt EB, Kersten A, Kong L, et al. Haloperidol use is associated with lower hospital mortality in mechanically ventilated patients. Crit Care Med. 2005;33:226–9.
Spronk PE, Riekerk B, Hofhuis J, et al. Occurrence of delirium is severely underestimated in the ICU during daily care. Intensive Care Med. 2009;35:1276–80.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, E.H.Z., Mabasa, V.H., Loh, G.W. et al. Haloperidol Dosing Strategies in the Treatment of Delirium in the Critically-Ill. Neurocrit Care 16, 170–183 (2012). https://doi.org/10.1007/s12028-011-9643-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-011-9643-3