
Abstract
The emergence of autoimmunity after vaccination has been described in many case reports and series. Everyday there is more evidence that this relationship is more than casual. In humans, adjuvants can induce non-specific constitutional, musculoskeletal or neurological clinical manifestations and in certain cases can lead to the appearance or acceleration of an autoimmune disease in a subject with genetic susceptibility. The fact that vaccines and adjuvants can trigger a pathogenic autoimmune response is corroborated by animal models. The use of animal models has enabled the study of the effects of application of adjuvants in a homogeneous population with certain genetic backgrounds. In some cases, adjuvants may trigger generalized autoimmune response, resulting in multiple auto-antibodies, but sometimes they can reproduce human autoimmune diseases including rheumatoid arthritis, systemic lupus erythematosus, Sjögren syndrome, autoimmune thyroiditis and antiphospholipid syndrome and may provide insights about the potential adverse effects of adjuvants. Likewise, they give information about the clinical, immunological and histologic characteristics of autoimmune diseases in many organs, especially secondary lymphoid tissue. Through the description of the physiopathological characteristics of autoimmune diseases reproduced in animal models, new treatment targets can be described and maybe in the future, we will be able to recognize some high-risk population in whom the avoidance of certain adjuvants can reduce the incidence of autoimmune diseases, which typically results in high morbidity and mortality in young people. Herein, we describe the main animal models that can reproduce human autoimmune diseases with emphasis in how they are similar to human conditions.
Similar content being viewed by others


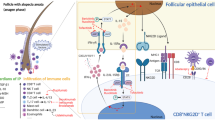
Introduction
The word adjuvant comes from the Latin adjuvare that means to help [1]. An immunological adjuvant is a substance that enhances a specific immune response without triggering any specific effect itself. The main uses of adjuvants include the enhancement of immunogenicity of highly purified or recombinant antigens that allow the reduction in the amount and number of immunizations needed for protective immunity, especially in certain populations such as newborns, elderly or immunosuppressed people. They also can be used as delivery systems for antigen uptake by mucosa and induce qualitative changes in the immune response, promoting a certain type of immune response not normally generated without adjuvant [1, 2].
Some proposed mechanisms by which adjuvants are able to increase the immune reaction include: delivering antigens to the immune system [2], activation of innate immune cells triggered by recognition of pathogen-associated molecular patterns by Toll-like receptors (TLR), NOD-like receptors (NOR), helicases (RIG-I-like receptors) and C-type lectin receptors [1] which are responsible for the induction of expression of co-stimulatory molecules [2]. Adjuvants can promote the attraction of dendritic cells toward the injection site [3], increase the uptake of the antigen by antigen-presenting cells [2], promote the secretion of cytokines with secondary activation of antigen-presenting cells (APCs) and favor the transport of antigen-loaded APCs toward the lymphoid tissue [2]. In addition, they allow progressive release, delayed clearance and better exposure of the antigen to the immune system [2].
Adjuvant classification
Adjuvants can be derived from many substances including oils, mineral salts, bacterial products (detoxified derivative monophosphoryl lipid A (MPL), lipopolysaccharide (LPS), peptidoglycan (PGN) [1]), xenobiotic (unmethylated CpG dinucleotides) and tuftsin auto-adjuvant [4–6].
According to Edelman’s classification, adjuvants can be subdivided in three groups [7]:
-
1.
Active immune stimulants, being substances that increase the immune response to the antigen [7].
-
2.
Carriers, being immunogenic proteins that provide T cell help [7].
-
3.
Vehicle adjuvants, being oil emulsions or liposomes that serve as a matrix for antigens as well as stimulating the immune response [7].
An alternative adjuvant classification divides adjuvants according to administration route (mucosal or parenteral). Another one divides them into alum salts and other mineral adjuvants, tensoactive agents, bacterial derivatives, vehicles and slow release materials or cytokines [7]. A fourth more recently proposed system of classification divides adjuvants into the following groups: gel-based adjuvants, tensoactive agents, bacterial products, oil emulsions, particulated adjuvants and fusion proteins or lipopeptides [7].
Main type of adjuvants used in human and veterinary vaccines and their relationship with inflammation and autoimmunity
Nowadays, most of the adjuvants used in human and veterinary vaccines were derived from oils and mineral salts.
Important examples of oil-based adjuvants include complete and incomplete Freund’s adjuvant (CFA and IFA, respectively). The former is a mixture of paraffin oil, surfactant and heat-killed Mycobacterium or PGN, LPS and N-acetyl muramyl-l-alanyl-d-isoglutamine (MDP), while the latter is the same as CFA but without microorganisms’ components [1, 5, 6].
Other oil-based adjuvants include pristane and squalene. Pristane is an isoprenoid alkaline found in mineral oils often contained in veterinary vaccines and has been shown to induce arthritis in susceptible mice strains [8]. It also can produce chronic inflammation when administered into the peritoneal cavity [5]. In addition, Anderson and Potter showed that i.p. injection of pristane into BALB/c mice could induce plasmacytoma 6–10 months after treatment [9]. In another experiment by Platica et al. [9], they inoculated pristane in the right paravertebral area with plasmacytoma cells. Pristane induced an intense local inflammatory response and produced an earlier and more frequent occurrence of plasmacytoma in mice in comparison with controls underlining the capacity of this adjuvant to promote hematologic tumors growth, probably through chronic inflammation (a similar mechanism as the induction of breast lymphoma after silicon implants) [5, 8–10].
Squalene (MF59) and squalene, α-tocopherol (AS03), are oil-based adjuvants which are known to increase uptake of antigens by antigen-presenting cells with consequent activation of local immune response and generation of B and T cell immunity with a mixed Th1 and Th2 cell phenotype without having systemic effects. Although it is considered to be safe in humans, squalene has been linked to Gulf war syndrome [6].
Nowadays, aluminum salts (alum) are the main type of adjuvants used in human vaccines. The adjuvant effect of alum implies trapping soluble antigens in the aluminum gel, interacting with dendritic cells enhancing antigen presentation, complement and eosinophil activation, as well as promoting an influx of neutrophils and enhancing the secretion of pro-inflammatory cytokines and chemokines. Alum can also induce cellular damage with liberation of intracellular DNA and uric acid, activating NALP3 inflammasome in macrophages with subsequent IL-1β secretion [5, 11].
In humans, immune responses to proteins with alum tend to be a mix of Th2 and Th1 cells; however, in mice alum induces a profoundly polarized Th2 cell response, with Th2 cell-dependent antibody isotypes, to nearly all protein antigens [1].
Aluminum hydroxide-containing vaccines include diphtheria, tetanus, pertussis and Haemophilus influenza [12]. Some studies have suggested the relationship between macrophagic myofasciitis (MMF) and chronic fatigue syndrome with vaccines containing aluminum, probably only in genetic-prone individuals such as those who carry the HLA-DRB1*01 haplotype [11]. Although of limited use in humans because of bacteria-derived adjuvants toxicity concerns, the work of Ribi et al. in 1982, led to the development of less toxic preparations of LPS, and ultimately to the substantially detoxified derivative monophosphoryl lipid A (MPL) [1]. MPL principally formulated with antigens and alum is now a component of licensed vaccines for hepatitis B and papilloma virus [1]. MPL formulated on alum (AS04) stimulates polarized Th1 cell response in contrast to the mixed Th1–Th2 cell response of alum alone [1]. Diseases associated with this adjuvant are hypothyroidism, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Sjögren syndrome (SS), dermatomyositis, erythema nodosum, Guillain Barré syndrome (GBS), optic neuritis, multiple sclerosis and myasthenia gravis [13].
Animal models of autoimmune diseases induced by adjuvants
Regardless of the benefits of adjuvants, they are not without side effects. Adverse reactions to adjuvants can be classified as local or systemic. Important local reactions include pain, local inflammation, swelling, injection site necrosis, lymphadenopathy, granulomas, ulcers and sterile abscess. Systemic reactions include nausea, fever, adjuvant arthritis, uveitis, eosinophilia, allergy, anaphylaxis, and organ-specific toxicity and immunotoxicity (i.e., the liberation of cytokines, and induction of immunosuppression or autoimmune diseases through molecular mimicry, bystander effects and non-specifically immune system stimulation [2, 5, 14, 15]). A vaccine-related autoimmune reaction should meet a series of requirements to be actually considered vaccine induced, including consistency, strength, specificity and temporal relation [16]. As shown in Table 1, many vaccines had been associated with human diseases [2, 16–21].
On the other hand, the adjuvant effect is known to be related to the autoimmune/auto-inflammatory syndrome induced by adjuvants (ASIA), which is the result of a hyperactive immune response after adjuvant exposure and encompasses silicosis, macrophagic myofasciitis, Gulf war syndrome and post-vaccination phenomena [22].
The suspected mechanism of adjuvant-induced autoimmunity include alteration of the host’s innate immune system through pathogen-associated molecular patterns receptors with subsequent cytokine production and bystander activation of potentially auto-reactive T cells that could bring help B cells leading to polyclonal activation. In addition, the interaction of certain adjuvants with self-proteins can induce conformational changes exposing certain epitopes that can be recognized by T cells (epitope spreading). Also, viral antigens with molecular mimicry with self-antigens can promote the production of auto-antibodies [23].
Animal models used in the study of autoimmune and inflammatory diseases can be broadly classified as either spontaneous, where an animal’s genetic background results in a defined prevalence of disease, or induced, where disease is precipitated by exposure to defined antigens, adjuvants or other experimental reagents [24]. Moreover, adjuvants were administered to an immune-prone animal. Favoino et al. [12] compared the effects of intraperitoneal CFA with adjuvants commercially available for human use (IFA, squalene, alum) combined with KLH in a lupus-prone animal model (New Zealand black/New Zealand white (BW) F1 mice). They found that in the IFA group, a proteinuria mean score of 1 was observed at week 17 and increased to 2 and 2.5 at weeks 27 and 28. Also, in this group proteinuria was significantly higher at weeks 17, 18 and 19 and at weeks 20 and 21 [12]. In the CFA group, proteinuria became detectable at week 24 and reached a score of 1 and 1.5 at weeks 27 and 28. Proteinuria showed a similar trend in the squalene and alum groups, becoming detectable at week 28 and increasing from week 29. In the untreated group, proteinuria became detectable at week 32 [12]. Interestingly, there were no significant differences in the titers of anti-ssDNA or anti-dsDNA in the adjuvant-treated groups compared with the untreated group, while anti-RNP/Sm antibodies were not detected in any of the mice in the squalene and alum groups and there was no change in the lymphocyte subpopulation distribution by flow cytometry [12]. Although these results can hardly be extrapolated to humans because of the use of a high dose and a different route of immunization, they showed that adjuvants can accelerate autoimmune disease in an adequate genetic background [12].
In laboratory animals, injections or topical application of mineral oils can initiate pathogenic autoimmune responses after following any of these three experiments [6]:
-
1.
With the oil alone injected subcutaneously or directly into pre-exposed lymph nodes or by transdermal administration, inducing an experimental arthritis in rats or SLE-like syndrome in mice [6].
-
2.
With the oil plus a second immunostimulant, for example components from mycobacterial cells walls. In rats, it can cause systemic inflammation that is very damaging to the joints but may also be manifest in other tissues to a lesser degree [6].
-
3.
With the oil plus a second immunostimulant and also a tissue antigen, for example thyroglobulin. This three-part combination is necessary to establish cell-mediated immunity in many targeted tissues [6]. The example of this type of model is experimental autoimmune thyroiditis [25, 26].
Finally, the most interesting way to generate an experimental autoimmune disease is by administering an adjuvant in a non-autoimmune animal. Herein, we summarize the main animal models of adjuvants-induced autoimmune diseases.
ASIA
In animals, after adjuvant exposure there are signs and symptoms that resemble human ASIA. An animal model of ASIA in commercial sheep was described by Lujan et al. In this protocol, they observed that ASIA occurred in commercial sheep after exposure to adjuvants contained in the blue tongue vaccine. The disease encompasses an acute phase occurring after 2–6 days of adjuvant exposure and is manifested as neurological manifestations that sometimes progress to seizures and prostration and correlated with the histopathological finding of loss of white matter and acute meningoencephalitis with exuberant perivascular inflammatory infiltration including lymphocytes, neutrophils, as well as macrophages containing PAS-positive intracytoplasmic bodies and inflammatory cells in a phenomenon called emperipolesis. This phase is mostly explained by the process of aluminum transport to the brain that leads to the secretion of pro-inflammatory cytokines. Most sheep recover after this phase, but some of them progress to the chronic phase that also may appear without a previous acute phase and is dependent on external stimuli being cold weather the most important. In this phase, aluminum is detected in blood and the sheep have neurological abnormalities, cachexia, anasarca, coma and death. They also showed peripheral nerve thickening that corresponded to edema and myxoid tissue deposit in the histopathological analysis as well as perineuritis and membranous glomerulonephritis [27].
Posteriorly, they induced the syndrome in sheep by the administration of vaccines adjuvanted with Al and thimerosal, and they found neurological manifestations consisting in alternating excitement and depression, depletion of fat deposits and slight increase in diameter of some peripheral nerves. Alum was detected in spinal cord by electron microscopy [27]. Therefore, ovine natural ASIA is a systemic autoimmune disease. Neurological manifestations of ASIA can be induced experimentally in sheep after adjuvant exposure.
On the other hand, adjuvants may also reproduce human autoimmune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Sjögren’s syndrome (SS) and antiphospholipid syndrome (APS) [5].
RA-like disease
Adjuvant-induced arthritis is an erosive, sterile, chronic, tissue-specific, diarthrodial arthritis, although systemic manifestations may occur. The severity and chronicity is associated with certain MHC class II haplotypes [15]. All models are T cell dependent, and since nude strains are resistant, the disease is ameliorated after treatment with anti-αβTcR monoclonal antibody (R73) and it can be transferred to naïve mice by lymph node or spleen T cells [8, 28, 29].
After intradermal administration of the adjuvant, it disseminate to lymph nodes, which is associated with a systemic inflammatory response with pro-inflammatory cytokines liberation and increment in serum acute phase reactants. When the adjuvant enters the lymph nodes, it activates antigen-presenting cells which may stimulate auto-reactive T cells able to respond to different antigens, including extracellular matrix, heat shock proteins, ribonucleoproteins, glucose 6-phosphate isomerase. These auto-reactive T cells secrete IFN-γ and TNF-α, inducing arthritis with different duration and severity according to distinct genetic background [6, 8, 28, 30].
Pristane-induced arthritis
The induction of arthritis by pristane was first described in 1974 [6]. Pristane-induced arthritis (PIA) by intraperitoneal injection, shares many histopathological and serological features with rheumatoid arthritis, and it resembles the human condition by the presence of chronic symmetrical involvement of peripheral joints, the presence of rheumatoid factor, destruction of cartilage and bone and dependence of specific immunity, in particular CD4+ T cells [8].
Using the PIA model in dark agouti (DA) rats, the arthritis develops 2 weeks after pristane injection [15]. An episode of severe and destructive arthritis in the peripheral joints follows and gradually subsides 3 weeks later [15]; however, starting at round 6–8 weeks after pristane injection, a chronic relapsing disease develops which can reach almost as high severity as during the first arthritic episode and does not subside [15]. In PIA, the paws are gradually deformed, without excessive new bone formation as observed in other adjuvant-induced arthritis such as M. tuberculosum [31]. Histopathological examination shows a deformed joint architecture. Synovial hyperplasia is the first morphological change, and joints with clinical arthritis show a prominent pannus tissue containing neutrophils and large macrophage-like cells [31]. By immunohistochemical analysis, there is a large number of MHC class II-expressing cells and a few activated T cells expressing the IL-2 receptor, as well as both CD4+ and CD8+ T cells [31]. The first signs of bone erosions are seen around day 15, usually starting at the subchondral, but in joints with severe arthritis, the erosions spread along the periosteal surface [31]. By day 122, the clinically affected joints are severely compromised by the erosions and cartilage is almost lost [31].
Although IgM rheumatoid factor production is T cell independent in PIA [32], the model is dependent on T cells [31].
Usually, there is an increase in serum levels of IL-6, and the cytokine is not detected in resistant E3 strain [31]. Susceptibility to PIA has been associated with increased agalactosyl-IgG levels mediated by IL-6 production (usually at day 14 after pristane injection, which corresponds with the onset of arthritis), while protection against PIA is mediated by Th2-associated cytokines produced after hsp65 pre-immunization [31, 33].
The development of PIA is associated with increased levels of antibodies and heightened in vitro T cell responses against mycobacterial hsp65 and mammalian hsp60. The level of expression of hsp60 in joints parallels susceptibility to arthritis [34]. Other antigens proposed to be a target for PIA are collagen II and COMP [8]. The study by Hoffman et al. showed that the heterogeneous nuclear ribonucleoprotein (hnRNP)-A2 also known as the RA33 antigen was a target for auto-antibodies in PIA in 4 strains of rats (DA.1F, DA.1U, DA.1 l and DA) and the levels of auto-antibodies correlated with disease severity during the acute phase [8]. Anti-RA33 abs peaked around day 12 immediately before the appearance of the first symptoms, declined thereafter and increased again during the acute phase [8]. Histologically, there was overexpression of hnRNP-A2 during the acute phase of PIA in the majority of synoviocytes of the infiltrating pannus tissue. Increased expression was also found in chondrocytes [8], and it was associated with increases in mRNA levels detected by real-time PCR [8]. Also, upon stimulation with natural or recombinant hnRNP-A2, a pronounced and dose-dependent IFN-γ secretion was seen in T cells cultures from rats with PIA [8] highlighting the role of T cell immunity in the disease.
The study by Holmberg et al. [35] showed that after the injection of pristane, there are many highly activated T CD4+ lymphocytes expressing ICAM-1, IL-2R (CD25), RT-1D (MHC II-DR) and RT-1B (MHC II-DQ), infiltrating the synovia of DA rats. In addition, PIA was transferred by the lymph nodes and spleen T cells from rats immunized with pristane in a dose- and time-dependent manner [35], and this was prevented by blocking RT-1D (MHC II-DR) and RT-1B (MHC II-DQ) with antibodies administered i.p. [35] which confirms that the disease is restricted to MHC. Also, the splenocytes from both normal and pristane-primed rats produced high amounts of IFN-γ and TNF-α, but not IL-4 when re-stimulated in vitro with concanavalin A (con A) [35]. Recently Morgan et al. [36] found antibodies against pristane extract, the chaperone protein BiP, joint extract, hsp, DNA and extracellular matrix components 6 months after pristane injection, suggesting that PIA is characterized by antibodies against a broad list of antigens relevant in RA [36].
Innate immune system also plays a role in PIA. In this regard, TLR deserves a special mention since polyl: C (a synthetic TLR3 agonist) aggravates PIA and when its expression is knock down in vivo, the clinical symptom and pathological manifestations are diminished [37]. Also, TLR3 can induce the production of IFN-β, IL-6, IL-8, chemokines such as CCL5, IP-10/CXCL10 and MMP-3, MMP-13, thymic stromal lymphopoietin and VEGF in fibroblast-like synoviocytes (FLS) [37]. As shown by Zhu et al. [37], pristane-primed T cells are capable to induce the up-regulation of TLR3 mRNA in FLS as well as the secretion of TNF-α, IL-17a and IFN-γ. TNF-α blockade with etanercept and IFN-γ blockade with the monoclonal antibody DB1 prevented the pristane-primed T cells-induced expression of TLR3 and IL-6 mRNA [37].
CFA-induced arthritis
CFA-induced arthritis was described by Carl Pearson in 1956 [6] and is a severe, acute disease in rats [38].
Arthritis is produced by a single intradermal injection of 0.1 mL of CFA in Sprague–Dawley rats at the base of the tail [39]. Also, in autoimmunity-prone mice such as MRL/MpJ-lpr/lpr (MRL-lpr), Ratkay et al. [40] demonstrated the induction of arthritis characterized by swelling and erythema of the hind legs in 67–83 % of mice treated with CFA. The mice immunized with CFA showed higher synovial inflammation and subsynovial hyperplasia, cartilage erosion, bone destruction and histologic score and higher titers of auto-antibodies [40].
IFA-induced arthritis
Mineral oil or synthetic types of oils can induce an erosive polyarthritis when injected intradermally into DA rats [41, 42] which was described by Sandra Kleinau et al. [6]. IFA is not a pure oil, but contains 85 % mineral oils (Bayol F) and 15 % emulsifier (Arlacel A) [15], and its intradermal administration induces transcription of mRNA for pro-inflammatory cytokines such as IFN-γ and TNF-α [43].
As shown by Kleinau S et al. [41], the course of oil-induced arthritis depends on the route of administration, since arthritis after percutaneous administration of IFA is generally mild and transient in comparison with the intradermal injection of the same oil. Furthermore, the localization of arthritis is often confined to the front paws, but may progress to be symmetrical and involve the hind paws [41]. The duration of clinically evident arthritis varies, but extended at the most to 9 days [41]. Histologically, it is characterized by infiltration of mononuclear cells of the synovia, abundance of infiltrating polymorphonuclear cells and mononuclear cells in the subsynovial and periarticular tissues with minimal bone and cartilage erosions [41].
Avridine-induced arthritis
As shown in the study by Vingsbo et al. [38], arthritis could be induced by a subcutaneous injection at the base of the tail of 150 μL of avridine (N,N-dioctadecyl-N’,N’-bis (2-hydroexyethyl) propanediamine (=CP20961 = avridine) solubilized in Freund’s incomplete adjuvant in pathogen-free rats at a concentration of 0, 10, 25 or 50 mg/mL. Different rat strains showed different susceptibility to the disease, ranging from the high susceptible strain (DA rats), less susceptible rats (LEW.1A, LEW.1D and LEW.1N rats) and resistant (E3 rats) [38]. The problem with this model is that arthritis is very severe and it is induced by a mixture of avridine and mineral oil which has arthritogenic properties by itself [31].
Squalene-induced arthritis
Another adjuvant oil is the endogenous cholesterol precursor squalene (C30H50) [44] which is administered by intradermal injection and, in similarity to other arthritogenic adjuvants, induces a T cell-mediated joint-specific inflammation in DA rats [29]. Squalene-induced arthritis develops in 100 % of DA rats, with no apparent sex-linked difference [45]. The mean day of onset is 13 days post-induction, and the first signs of arthritis appear symmetrically in ankles and metatarsal joints of all paws and progress to include larger joint areas and finger joints [45]. Histopathological evaluation reveals a hypertrophic synovial tissue with pannus invading the joint space, osteolysis and chondrolysis. Neutrophils and αβ+ T cells are abundant within the joints and surrounding tissues [45]. After maximum score is reached, the joint inflammation gradually subsides and leaves few or no signs of macroscopic damage or ankylosis [45]. In the histologic analysis by immunohistochemistry, there is an up-regulation of CD4, CD8, αβ TcR, CD11b/c, MHC class II and PCNA (early cell activation) [45]. The inflammation is joint specific, since none of these markers are found in other tissues [45].
In genetically prone different rat strains, squalene-induced arthritis resembles rheumatoid arthritis since it is characterized by long-lasting symmetrical arthritis, involves at least three groups of joints including hand joints and produces bone erosions [44].
The joint-restricted inflammation in this model results from the permanence of squalene at the injection site and lymph nodes [29]. Holm et al. [29] compared the production of pro-inflammatory cytokines by T cells from the lymph nodes draining the sites of squalene injection, and they found a stronger expression of IL-1β and IFN-γ after the injection of squalene before and after the development of arthritis. There was an important Th1 response manifested as higher IFN-γ/IL-4 ratio in draining lymph nodes. The disease was transferred to naïve rats by the administration of T cells from draining and non-draining lymph nodes from squalene-treated rats, although the latter was milder [29].
In summary, PIA reproduces most of the clinical characteristics of rheumatoid arthritis, which makes it an attractive model to study new therapeutic targets.
SLE-like disease
Alum- and HBVv-induced lupus
Vaccines can accelerate SLE in a lupus-prone animal. In the experiment by Agmon-Levine et al, three groups of 20 NZBWF1 mice each were inoculated intramuscularly with PBS or the hepatitis B virus (HBV) vaccine (Engerix-B vaccine) and aluminum hydroxide in a quantity comparable with that contained in the vaccine at the ages of 8 and 12 weeks. Mice weight was measured monthly, and they took blood samples and analyzed urine protein at different moments. Neuro-cognitive tests were performed at 21, 26 and 30 weeks of age. The mice inoculated with alum showed a lower hematocrit, white and red blood cells count in comparison with mice inoculated with PBS. Mice inoculated with HBVv showed lower red blood cells count and higher anti-dsDNA antibody levels in comparison with mice inoculated with PBS. The urine protein levels were higher in mice inoculated with HBVv in comparison with PBS and alum. All mice presented SLE-like kidney damage, but those inoculated with HBVv showed crescent proliferative glomerulonephritis, while the other groups showed mesangial disease with less Ig deposits. The hepatitis B surface antigen was detected in the kidney of the animals immunized with the vaccine [14]. Mice immunized with alum and HBVv showed short and long memory shortages. Alum-inoculated mice were found to be more anxious, and there was an increment in the number of astrocytes and alum deposition in the brain ventricle [14].
Pristane-induced lupus
Pristane is able to induce the production of lupus autoantibodies in practically any genetic background although at different frequencies, as shown by Satoh et al. In their study, they found that anti-nRNP/Sm and anti-Su autoantibodies were produced by A.SW (H-2s), B6 (H-2d) and DBA/1 (H-2q) mice, whereas antiribosomal P antibodies were produced by A.SW and B6 mice but not by BALB/c or DBA/1 [46]. BALB/c, SJL/j and C57BL/6 mice injected intraperitoneally with pristane develop lupus-specific auto-antibodies, including anti-nRNP/Sm and ribosomal P, as well as the less disease-specific auto-antibodies anti-Su, histone and ssDNA with severe immune complex-mediated glomerulonephritis characterized by segmental or diffuse proliferative lesions with IgG1, IgG2a, IgG2b and IgG3 glomerular deposit starting 4–6 months after pristane treatment [32, 47, 48]. This model is related to lymphoid neogenesis, the formation of ectopic lymphoid tissue that closely resembles secondary lymphoid tissue and arises by similar developmental pathway at sites of inflammation [32]. In fact, IgM and IgG antibody-forming cells producing antibodies to U1-A protein, a component of U1 snRNPs, are more abundant in the ectopic lymphoid tissue than in the spleen [32]. Likewise, this tissue is an important source of type I IFN and shows increased expression of IFN-stimulated genes (ISGs) including Mx-1, IRF-7, ISG15 and IP-10 [32]. Therefore, this is the only animal model capable to reproduce the IFN signature which is also seen in peripheral blood of pristane-treated wild-type mice and is dependent on the TLR7-Myd88 signaling pathway [32]. Although the activation mechanism is undefined, pristane might augment the response to endogenous TLR7 ligands such as the U1 RNA component of Sm or RNP antigen. Also, incorporation of pristane into the cell membrane might disturb the endosomal location of TLR7, providing access to endogenous ligands [32]. In addition, pristane might interfere with degradation of cellular debris, increasing the availability of endogenous nucleic acids [32].
Moreover, the ability of pristane to promote auto-antibody production requires Fas–Fas ligand signaling since both B6-lpr/lpr (Fas deficient) and B6-gld/gld (Fas ligand deficient) mice are highly resistant, raising the possibility that Fas-mediated apoptosis might generate endogenous TLR7 ligands [32] with subsequent auto-antibodies production. Thus, pristane-induced lupus reproduces the IFN signature, and type I IFN is produced by inflammatory monocytes newly released from the bone marrow and expressing high levels of TLR7 and Ly6C (Ly6Chi monocytes) [32]. Ly6Chi monocytes are attracted to the peritoneal cavity after pristane injection by CCL2 and also accumulate in ectopic lymphoid tissue induced by pristane [32].
As shown by Feng et al. [49], irf5 −/− mC57B1/6 mice have diminished production of highly pathogenic IgG2a/c and IgG2b antibodies and have an increase in the less pathogenic IgG1-specific lupus auto-antibodies, and the effect persists after 6 months of pristane treatment. Also, irf5 −/− mC57B1/6 mice showed polarization to Th2 profile with IL-4 secretion that promote production of the less pathogenic IgG1 isotype and had diminished IL-6 production [49]. In addition, irf5 −/− mC57B1/6 mice lose the type I IFN signature since a significant decrease in mRNA levels of the interferon-stimulated gene IRF7 was detected in L929 cells stimulated sera from pristane-injected Irf5 −/− mice. There is no increase in surface expression of the ISG Sca-1 in CD19+ B cells from peripheral blood of Irf5 −/− mice 2 weeks post-pristane injection. Similar results were found at 6 months post-injection in different cellular compartments of Irf5 −/− mice, and decreased mRNA expression of the ISGs MCP-1 (ccl2) and MX1 (myxoma response protein) was also observed in the bone marrow [49]. IFN-deficient mice are resistant to the induction of IgG antichromatin autoantibodies, but still produce IgG2a anti-Sm or RNP [32]. Type I IFN signaling is essential for anti-Sm or RNP autoantibodies and also promotes switching to IgG2a [32]. Along with type I IFNs, IL-6, IFNγ and IL-12 promote autoantibody production in pristane-induced lupus.
IL-6 deficiency abrogates the induction of IgG anti-DNA and chromatin, but not anti-Sm, RNP or Su auto-antibodies [32]. Richards et al. [47] showed that ssDNA antibodies can be produced in BALBc mice immunized with pristane in a IL-6-independent manner since the antibodies were found in similar titers in IL-6−/− versus IL-6+/+ mice 3 weeks after pristane treatment. In contrast, IgG anti-ssDNA antibodies were detected at a high frequency 8 months after pristane treatment only in IL-6+/+ mice (p < 0.05) [47]. In this study, it was shown for the first time that anti-dsDNA antibodies could be detected by Crithidia luciliae kinetoplast staining assay and ELISA in a specific strain of mouse (BALB/cAn (IL-6 +/+) 8 months after pristane treatment [47]. These results suggest that BALB/cAn mice can develop anti-dsDNA antibodies in an IL-6-dependent manner [47]. Also IL-6 is important in pristane-induced antichromatin auto-antibodies production [47].
The auto-antibodies in pristane-induced lupus do not completely develop in BALB/c nu/nu (nude) mice or T cell receptor-deficient (C57BL6 TcRβ −/−, TcRδ −/−) mice following pristane treatment [32]. Thus, auto-antibodies repertoire is determined by the presence of T cells, as shown by Richards et al. [50]. They found that nude BALB/c ByJ mice (BALB/c ByJ nu/nu) spontaneously produce IgM and IgG anti-ssDNA and chromatin antibodies. Pristane treatment enhanced the production of IgM anti-ssDNA and IgG antichromatin autoantibodies in nu/nu mice, suggesting a T cell-independent response [50]. In contrast, neither anti-nRNP/Sm nor anti-Su antibodies were detected in nu/nu mice, so these auto-antibodies are T cell dependent [50]. Also in the histopathological analysis, pristane-treated nu/+ mice had a higher frequency of IgG2a, IgG2b and C3 deposits than similarly treated nu/nu mice (p < 0.05), suggesting that T cells are necessary for the production of these more pathogenic auto-antibodies [50].
Pristane induces immune complex-mediated glomerulonephritis mediated by IL-6, IFNα/β, IFNγ and IL-12p35 and able to resemble lupus nephritis as well as alveolar hemorrhage and TNF-α-mediated arthritis [32, 51]. Another key cytokine that recently has been related to lupus pathogenesis is IL-17. As shown by Summer et al. [52], C57BL/6 mice treated with pristane have increased number of IL-17A-producing peritoneal macrophages (F4/80+) and neutrophils (Ly6G+) in comparison with controls found by flow cytometry. Also, their splenocytes have increased the production of IL-17A, and this is enhanced when they are stimulated with TLR4 and TLR2 ligands [52]. The production of the Th1 cytokine IFN-γ, TNF, anti-dsDNA antibodies and anti-RNP antibodies was diminished in IL-17A−/− mice, and they had less IgG and C3 deposit in the glomeruli [52]. IL-17A−/− mice had less CD4+ glomerular T cells, developed only modest proteinuria and had attenuated histological renal injury 7 months after pristane injection, in comparison with the WT, which became proteinuric at 3 months after pristane treatment [52].
The pristane-induced immune complex-mediated proliferative glomerulonephritis [53] is more severe in BALB/c mice lacking FcγRIIB receptor. They show more prominent glomerular IgG deposits, increased sclerosis and crescent formation. This may be because FcγRIIB acts to maintain tolerance by limiting B cell antigen receptor activation and deleting auto-reactive lymphocytes that might arise during somatic hypermutation [53]. Thus, loss of inhibitory FcγR regulation by FcγRIIB receptor increases susceptibility to pristane-induced nephritis [53]. Interestingly, in FcγRI/III−/− mice, proteinuria was abrogated despite the persistence of deposition of glomerular IgG and complement [53]. Although FcγRIIB−/− mice spontaneously develop polyclonal hypergammaglobulinemia and antichromatin antibodies, pristane treatment enhances both total IgG and antichromatin production, but not the spontaneous production of anti-dsDNA, anti-Su or anti-nRNP/Sm antibodies [53]. IL-6 is elevated in the supernatant of the FcγRIIB−/− peritoneal cells after the stimulation with anti-CD3, lipopolysaccharide or Con A, and pristane enhances its production and the secretion of other cytokines such as IL-12, IFN-γ and TNF-α in a Fcγ-independent manner since the levels of cytokines were equal in FcγRIIB−/−, RI/RIII−/− and +/+ mice [53].
Squalene-induced lupus
IFA and squalene induce similar spectrum of autoantibodies, but less efficiently than pristane [32].
In summary, pristane-induced lupus reproduces most of the immunological characteristics of the disease, including the systemic autoimmunity and IFN signature. Pristane is able to induce lupus even in non-autoimmune-prone mice, which highlights the importance of adjuvants as triggers of autoimmune diseases.
Sjögren syndrome-like disease
In genetically prone mice, adjuvants can induce SS, as shown in the next model.
Alum-induced SS
SS can be induced in an animal model of female NZM2758 mice. This mice strain is generated by in-breeding of the NZB and NZW mice and is genetically susceptible to SS. The mice develop reduced salivary gland function and sialadenitis following injection of IFA [54]. Another way to induce SS in this mouse strain is by subcutaneous injection of 50 % alum suspension mixed with PBS with additional intraperitoneal injections of alum (0.05 mL/mouse) 4 and 8 weeks later. Pilocarpine-induced saliva production was measured at 8 weeks after the first injection. Anti-nuclear antibodies, as well as antibodies to Ro60 and La were measured. The injection of alum induced a significant drop in the mean saliva volume, and this effect persisted over time until 20 weeks, when experiment was terminated. In the histological analysis of salivary glands, all mice showed multiple inflammatory foci in the peri-vascular or peri-ductal regions and smaller foci in the surrounding secretory acini. The total inflammation score was higher in the alum-treated mice (p = 0.012). One of the suggested mechanisms of SS induction in these mice is activation of the P2X7 receptor (P2X7R) in the salivary glands with concomitant increase in the gene expression of inflammasome components (NLRP3, ASC and caspase-1) with subsequent release of pro-inflammatory cytokines such as IL-1β and IL-18 which are known to be high in patients with SS [54].
In conclusion, an adjuvant like alum was able to worsen the clinical manifestations of SS, proving the importance of environmental factors as triggers of autoimmune symptoms.
Autoimmune thyroid disease-like disease
Experimental autoimmune thyroiditis (EAT) in mice is linked to certain haplotypes of the mouse major histocompatibility complex, the H-2 complex [55]. EAT is characterized by both antibody production to the inducing auto-antigen (mouse thyroglobulin (MTg)) and infiltration of the thyroid by mononuclear cells including lymphocytes and macrophages [55]. The disease is only induced in the presence of T cells from high-responder susceptible strains [55]. The immune response gene (Ir-Tg) was mapped to the I–A subregion [55]. In susceptible strains, both CFA and bacterial lipopolysaccharide (LPS) serve as potent adjuvants for MTg [55]. The injection of purified thyroglobulin plus CFA induces production of both thyroglobulin-specific autoantibodies and inflammatory lesions of the thyroid gland in contrast with IFA that gave rise to autoantibody production but no lesions in the thyroid gland [25]. On the other hand, injection of thyroglobulin and concurrent intravenous administration of bacterial LPS induces the full picture of autoimmune disease with the production of both autoantibodies and lesions in the thyroid [25].
APS-like disease
In APS, anti-β2GPI antibodies are part of the disease classification criteria and represent a risk factor for thrombosis. As shown by professor Blank, the β2GPI has a peptide in its domain number 3 (TLRVYK) that is present in many infectious agents including tetanus toxoid (TTd). After TTd immunization, mice produce anti-β2GPI and antipeptide (TLRVYK) antibodies [56]. When the antipeptide (TLRVYK) antibody was purified and administered to naïve mice, they developed the clinical manifestations of APS (fetal loss, thrombocytopenia and prolonged PTT) [56]. Therefore, tetanus toxoid hyperimmunization is a suitable model of experimental APS in mice. The model is dependent on different genetic background since BALB/c and C57BL/6 have distinct reproductive pathology. But also, the model is variable according to the type of administered adjuvant and the titers and specificity of anti-β2GPI antibodies. Indeed, pretreatment with CFA leads to the production of high-affinity anti-β2GPI antibodies through molecular mimicry. The effect was reduction in fecundity without affecting fertility in C57BL/6 mice and reduction in fertility without affecting fecundity in BALB/c mice. Treatment with LPS and TTd after CFA pretreatment leads to a reduction in low-affinity antibodies that are known to be natural and protective, leading to decreased fecundity and fertility, while administration of TTd with PGN induced lower fertility, fecundity and smaller pups, related to augmentation of low-affinity antibodies [57–59], highlighting the importance of the interactions between adjuvants and a genetics in the induction of a pathogenic autoimmune response.
In summary, TTd-induced APS is an example of infections and vaccines as triggers of autoimmune diseases through molecular mimicry.
Granuloma in salmon fish
Finally, the i.p. administration of different oily adjuvants is related to polyclonal B cell activation, leading to the production of antinuclear antibodies with many different patterns and antibodies against distinct auto-antigens, but also a chronic hepatic and peritoneal granulomatous inflammation, with portal vein thrombosis, focal immune complex-mediated endocapillary proliferative GMN and thickening of glomerural basal membrane by electron microscopy [60].
In summary, animal models prove the fact that adjuvants can cause autoimmune diseases in autoimmune- and non-autoimmune-prone subjects even without co-administration of a specific antigen. Animal models would help us unveil genetic traits related to adjuvants- and vaccines-induced autoimmunity. Maybe in the future, that may enable us to avoid certain vaccines and adjuvants in susceptible individuals or to use different routs of administration that may lead to personalized vaccines.
References
Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33(4):492–503.
Ahmed SS, Schur PH, Macdonald NE, Steinman L, Narcolepsy. A(H1N1) pandemic influenza, and pandemic influenza vaccinations: what is known and unknown about the neurological disorder, the role for autoimmunity, and vaccine adjuvants. J Autoimmun. 2014;50:1–11.
Fournie GJ, Mas M, Cautain B, et al. Induction of autoimmunity through bystander effects. Lessons from immunological disorders induced by heavy metals. J Autoimmun. 2001;16(3):319–26.
Guimaraes LE, Baker B, Perricone C, Shoenfeld Y. Vaccines, adjuvants and autoimmunity. Pharmacol Res. 2015;100:190–209.
Cruz-Tapias P, Agmon-Levin N, Israeli E, Anaya JM, Shoenfeld Y. Autoimmune (auto-inflammatory) syndrome induced by adjuvants (ASIA)—animal models as a proof of concept. Curr Med Chem. 2013;20:4030–6.
Whitehouse M. Oily adjuvants and autoimmunity: Now time for reconsideration? Lupus. 2012;21:217–22.
Petrovsky N, Aguilar JC. Vaccine adjuvants: current state and future trends. Immunol Cell Biol. 2004;82:9.
Hoffmann MH, Tuncel J, Skriner K, et al. The rheumatoid arthritis-associated autoantigen hnRNP-A2 (RA33) is a major stimulator of autoimmunity in rats with pristane-induced arthritis. J Immunol. 2007;179(11):7568–76.
Platica M, Bojko C, Steiner G, Hollander VP. Effect of subcutaneously administered 2,6,10,14-tetramethylpentadecane on plasmacytoma growth. Cancer Res. 1980;40:2229–33.
Bizjak M, Selmi C, Praprotnik S, et al. Silicone implants and lymphoma: the role of inflammation. J Autoimmun. 2015;65:64–73.
Israeli E, Agmon-Levin N, Blank M, Shoenfeld Y. Macrophagic myofaciitis a vaccine (alum) autoimmune-related disease. Clin Rev Allergy Immunol. 2011;41(2):163–8.
Favoino E, Favia EI, Digiglio L, Racanelli V, Shoenfeld Y, Perosa F. Effects of adjuvants for human use in systemic lupus erythematosus (SLE)-prone (New Zealand black/New Zealand white) F1 mice. Clin Exp Immunol. 2012;175:32–40.
Soldevilla HF, Briones SFR, Navarra SV. Systemic lupus erythematosus following HPV immunization or infection? Lupus. 2012;21:4.
Agmon-Levin N, Arango MT, Kivity S, et al. Immunization with hepatitis B vaccine accelerates SLE-like disease in a murine model. J Autoimmun. 2014;54:21–32.
Holmdahl R, Lorentzen JC, Lu S, et al. Arthritis induced in rats with non-immunogenic adjuvants as models for rheumatoid arthritis. Immunol Rev. 2001;184:19.
Salemo S, D’Amelio R. Could autoimmunity be induced by vaccination? Int Rev Immunol. 2010;29:247–69.
Stratton KR, Howe CJ, Johnston RB Jr. Adverse events associated with childhood vaccines other than pertussis and rubella. Summary of a report from the Institute of Medicine. JAMA. 1994;271(20):1602–5.
Cohen AD, Shoenfeld Y. Vaccine-induced autoimmunity. J Autoimmun. 1996;9(6):699–703.
Ropper AH, Victor M. Influenza vaccination and the Guillain–Barré syndrome. N Engl J Med. 1998;339(25):1845–6.
Tishler M, Shoenfeld Y. Vaccination may be associated with autoimmune diseases. Isr Med Assoc J. 2004;6(7):430–2.
Praprotnik S, Sodin-Semrl S, Tomsic M, Shoenfeld Y. The curiously suspicious: infectious disease may ameliorate an ongoing autoimmune destruction in systemic lupus erythematosus patients. J Autoimmun. 2008;30(1–2):37–41.
Shoenfeld Y, Agmon-Levin N. ASIA Autoimmune/inflammatory syndrome induced by adjuvants. J Autoimmun. 2011;36(1):4–8.
Perricone C, Colafrancesco S, Mazor RD, Soriano A, Agmon-Levin N, Shoenfeld Y. Autoimmune/inflammatory syndrome induced by adjuvants (ASIA) 2013: unveiling the pathogenic, clinical and diagnostic aspects. J Autoimmun. 2013;47:1–16.
Hall SW, Cooke A. Autoimmunity and inflammation: murine models and translational studies. Mamm Genome. 2011;22(7–8):377–89.
Rose NR. The adjuvant effect in infection and autoimmunity. Clin Rev Allergy Immunol. 2008;34(3):279–82.
Vladutiu AO, Rose N. Autoimmune murine thyroiditis relation to histocompatibility (H-2) type. Science. 1971;174(4014):1137–9.
Lujan L, Pérez M, Salazar E, et al. Autoimmune/autoinflammatory syndrome induced by adjuvants (ASIA syndrome) in commercial sheep. Immunol Res. 2013;56(2–3):317–24.
Wester L, Olofsson P, Ibrahim SM, Holmdahl R. Chronicity of pristane-induced arthritis in rats is controlled by genes on chromosome 14. J Autoimmun. 2003;21(4):305–13.
Holm BC, Lorentzen JC, Bucht A. Adjuvant oil induces waves of arthritogenic lymph node cells prior to arthritis onset. Clin Exp Immunol. 2004;137(1):59–64.
Wester L, Koczan D, Holmberg J, et al. Differential gene expression in pristane-induced arthritis susceptible DA versus resistant E3 rats. Arthritis Res Ther. 2003;5(6):R361–72.
Vingsbo C, Sahlstrand P, Brun JG, Jonsson R, Saxne T, Holmdahl R. Pristane-induced arthritis in rats a new model for rheumatoid arthritis with a chronic disease course influenced by both major histocompatibility complex and non-major histocompatibility complex genes. Am J Pathol. 1996;149(5):9.
Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30(9):455–64.
De Franco M, Peters LC, Correa MA, et al. Pristane-induced arthritis loci interact with the Slc11a1 gene to determine susceptibility in mice selected for high inflammation. PLoS ONE. 2014;9(2):e88302.
Barker RN, Easterfield AJ, Allen RF, Wells AD, Elson CJ, Thompson SJ. B- and T-cell autoantigens in pristane-induced arthritis. Immunology. 1996;89:6.
Holmberg J, Tuncel J, Yamada H, Lu S, Olofsson P, Holmdahl R. Pristane, a Non-antigenic adjuvant, induces MHC class II-restricted, arthritogenic T cells in the rat. J Immunol. 2006;176(2):1172–9.
Morgan R, Wu B, Song Z, Wooley PH. Immune reactivity to connective tissue antigens in pristane induced arthritis. J Rheumatol. 2004;31:10.
Zhu W, Jiang C, Xu J, et al. Pristane primed rat T cells enhance TLR3 expression of fibroblast-like synoviocytes via TNF-alpha initiated p38 MAPK and NF-kappaB pathways. Clin Immunol. 2015;156(2):141–53.
Vingsbo C, Sahistrand P, Brun JG, Jonsson R, Saxne T, Holmdahl R. Avridine-induced arthritis in rats; a T cell-dependent chronic disease influenced both by MHC genes and by non-MHC genes. Clin Exp Immunol. 1995;99(3):359–63.
Kaibara N, Hotokebuchi T, Takagishi K, et al. Pathogenic difference between collagen arthritis and adjuvant arthritis. J Exp Med. 1984;159:9.
Ratkay LG, Zhang L, Tonzetich J, Waterfield JD. Complete Freund’s adjuvant induces an earlier and more severe arthritis in MRL-ipr mice. J Immunol. 1993;151(1):5081–7.
Kleinau S, Erlandsson H, Klareskog L. Percutaneous exposure of adjuvant oil causes arthritis in DA rats. Clin Exp Immunol. 1994;96(2):281–4.
Svelander L, Müssener A, Erlandsson-Harris H, Kleinau S. Polyclonal Thl cells transfer oil-induced arthritis. Immunology. 1994;91(2):260–5.
Lorentzen JC, Glaser A, Jacobsson L, et al. Identification of rat susceptibility loci for adjuvant-oil-induced arthritis. Proc Natl Acad Sci USA. 1998;95:5.
Holm BC, Xu HW, Jacobsson L, Larsson A, Luthman H, Lorentzen J. Rats made congenic for Oia3 on chromosome 10 become susceptible to squalene-induced arthritis. Hum Mol Gen. 2001;10(6):565–72.
Carlson BC, Jansson AM, Larsson A, Bucht A, Lorentzen JC. The endogenous adjuvant squalene can induce a chronic T-cell-mediated arthritis in rats. Am J Pathol. 2000;156:2057–65.
Satoh M, Richards HB, Shaheen VM, et al. Widespread susceptibility among inbred mouse strains to the induction of lupus autoantibodies by pristane. Clin Exp Immunol. 2000;121:399–405.
Richards HB, Satoh M, Shaw M, Libert C, Poli V, Reeves WH. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J Exp Med. 1998;188(5):6.
Satoh M, Kumar A, Kanwar YS, Reeves WH. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proc Natl Acad Sci USA. 1995;92(24):10934–8.
Feng D, Yang L, Bi X, Stone RC, Patel P, Barnes BJ. Protection of Irf5-deficient mice from pristane-induced lupus involves altered cytokine production and class switching. Eur J Immunol. 2012;42(6):1477–87.
Richards HB, Satoh M, Jennette JC, Okano T, Kanwar YS, Reeves WH. Disparate T cell requirements of two subsets of lupus-specific autoantibodies in pristane-treated mice. Clin Exp Immunol. 1999;115(3):547–53.
Perry D, Sang A, Yin Y, Zheng YY, Morel L. Murine models of systemic lupus erythematosus. J Biomed Biotechnol. 2011;2011:1–19.
Summers SA, Odobasic D, Khouri MB, et al. Endogenous interleukin (IL)-17A promotes pristane-induced systemic autoimmunity and lupus nephritis induced by pristane. Clin Exp Immunol. 2014;176:342–50.
Clynes R, Calvani N, Croker PB, Richards HB. Modulation of the immune response in pristane-induced lupus by expression of activation and inhibitory Fc receptors. Clin Exp Immunol. 2005;141:230–7.
Bagavant H, Nandula SR, Kaplonek P, Rybakowska PD, Deshmukh US. Alum, an aluminium based adjuvant, induces Sjögren’s syndrome-like disorder in mice. Clin Exp Rheumatol. 2014;32(2):251–5.
Kong YC, Audibert F, Giraldo AA, Rose NR, Chedid L. Effects of natural or synthetic microbial adjuvants on induction of autoimmune thyroiditis. Infect Immun. 1985;49:40–5.
Blank M, Krause I, Fridkin M, et al. Bacterial induction of autoantibodies to β2-glycoprotein-I accounts for the infectious etiology of antiphospholipid syndrome. J Clin Invest. 2002;109(6):797–804.
Zivkovic I, Stojanovic M, Petrusic V, Inic-Kanada A, Dimitrijevic L. Induction of APS after TTd hyper-immunization has a different outcome in BALB/c and C57BL/6 mice. Am J Reprod Immunol. 2011;65(5):492–502.
Petrusic V, Zivkovic I, Muhandes L, Dimitrijevic R, Stojanovic M, Dimitrijevic L. Infection-induced autoantibodies and pregnancy related pathology: an animal model. Reprod Fertil Dev. 2014;26(4):578–86.
Petrusic V, Todorovic N, Zivkovic I, et al. Autoantibody response and pregnancy-related pathology induced by combined LPS and tetanus toxoid hyperimmunization in BALB/c and C57BL/6 mice. Autoimmunity. 2015;48(2):87–99.
Koppang EO, Bjerkas I, Haugarvoll E, et al. Vaccination-induced systemic autoimmunity in farmed Atlantic salmon. J Immunol. 2008;181(7):4807–14.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ruiz, J.T., Luján, L., Blank, M. et al. Adjuvants- and vaccines-induced autoimmunity: animal models. Immunol Res 65, 55–65 (2017). https://doi.org/10.1007/s12026-016-8819-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-016-8819-5