
Abstract
Aim
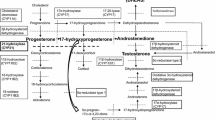
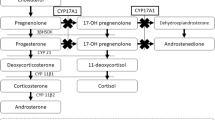
17α-hydroxylase enzyme deficiency is a rare form of congenital adrenal hyperplasia (CAH) and is caused by mutations in the CYP17A1 gene. The main clinical findings are delayed puberty and primary amenorrhea in girls, and disorders of sex development in boys. It can also cause hypertension and hypokalemia in both genders. In this study, we aimed to present the clinical, laboratory and genetic results of 13 patients from eight different families who were diagnosed with complete 17α-hydroxylase enzyme deficiency.
Methods
The age, symptoms, anthropometric measurements, blood pressure, Tanner stages, and hormonal and chromosome analysis results at the time of admission were recorded from the medical records of the patients. Whole gene next-generation sequencing of CYP17A1 gene was performed to detect mutations. Multiplex ligation dependent probe amplification (MLPA) method were used to detect deletions in the seven patients who had no point mutation were detected in the CYP17A1 gene.
Results
The average age of the patients at the time of admission was 14.8 (range: 12.9–16.6) years. Also at this time, all patients were in adolescence and were raised as females. The karyotypes of eight patients were 46,XY, and of five patients were 46,XX. Ten patients presented with delayed puberty and primary amenorrhea, one patient with delayed puberty and hypertension, and two patients with hypertension and/or hypokalemia. Hypertension and hypokalemia were detected in nine and seven patients, respectively.
Conclusions
P450c17 enzyme deficiency should be considered in patients presenting with delayed puberty or primary amenorrhea in the adolescence period and diagnosed with hypergonadotropic hypogonadism, if hypertension and hypokalemia accompany. Early diagnosis prevents the occurrence of important health problems such as hypertension, psychological problems, and gender identity disorders, which affect the majority of these patients.
Similar content being viewed by others
Data availability
Data is obtained from patient files.
References
W.L. Miller, R.J. Auchus, The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32(1), 81–151 (2011)
Y.K. Oh, U. Ryoo, D. Kim et al. 17a-hydroxlyase/17, 20-lyase deficiency in three siblings with primary amenorrhea and absence of secondary sexual development. J. Pediatr. Adolesc. Gynecol. 25, e103–e105 (2012)
S. Lieberman, P.A. Warne, 17-Hydroxylase: an evaluation of the present view of its catalytic role in steroidogenesis. J. Steroid Biochem. Mol. Biol. 78(4), 299–312 (2001)
M. Costa-Santos, C.E. Kater, R.J. Auchus; Brazilian Congenital Adrenal Hyperplasia Multicenter Study Group, Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 89, 49–60 (2004)
K. Miura, K. Yasuda, T. Yanase, N. Yamakita, H. Sasano, H. Nawata, M. Inoue, T. Fukaya, Y. Shizuta, Mutation of cytochrome P-45017 a gene (CYP17) in a Japanese patient previously reported as having glucocorticoid-responsive hyperaldosteronism: with a review of Japanese patients with mutations of CYP17. J. Clin. Endocrinol. Metab. 81, 3797–3801 (1996)
M. Zhang, S. Sun, Y. Liu, H. Zhang, Y. Jiao, W. Wang, X. Li, New, recurrent, and prevalent mutations: Clinical and molecular characterization of 26 Chinese patients with 17 alpha hydroxylase/17,20-lyase deficiency. J. Steroid Biochem Mol. Biol. 150, 11–16 (2015)
B. Han, L. Xue, M. Fan, S. Zhao, W. Liu, H. Zhu, T. Cheng, Y. Lu, K. Cheng, H. Song, Y. Liu, J. Qiao, Clinical and molecular manifestation of fifteen 17OHD patients: a novel mutation and a founder effect. Endocrine 53, 784–790 (2016)
A.D. Kardelen, G. Toksoy, F. Baş, Z.Y. Abalı, G. Gençay, Ş. Poyrazoğlu, R. Bundak, U. Altunoğlu, Ş. Avcı, A. Najaflı, O. Uyguner, B. Karaman, S. Başaran, F. Darendeliler, A rare cause of congenital adrenal hyperplasia: clinical and genetic findings and follow-up characteristics of six patients with 17-hydroxylase deficiency including two novel mutations. J. Clin. Res. Pediatr. Endocrinol. 10(3), 206–215 (2018)
F. Hannah-Shmouni, W. Chen, D.P. Merke, Genetics of congenital adrenal hyperplasia. Endocrinol. Metab. Clin. North Am. 46, 435–458 (2017)
M. Grumbach, I. Hughes, F. Conte. Williams textbook of endocrinology. Williams Textbook of Endocrinology. P.R. Larsen, H.M. Kronenb, S. Melmed editors. Saunders, Philadelphia), 2003) 842–1002
R.J. Auchus, Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J. Steroid. Biochem. Mol. Biol. 165, 71–78 (2017).
E. Unal, R. Yıldırım, F.F. Taş, S. Tekin, S. Ceylaner, Y.K. Haspolat, A rare cause of delayed puberty in two cases with 46,XX and 46,XY karyotype: 17 α-hydroxylase deficiency due to a novel variant in CYP17A1 gene. Gynecol. Endocrinol. 36(8), 739–742 (2020)
S. Bolu, R. Eröz, M. Tekin, M. Doğan, Atypical presentation in patients with 17 α-hydroxylase deficiency caused by a deletion in the CYP17A1 gene: short stature. Turk. J. Pediatr. 62, 851–857 (2020)
National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents, The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics 114(Suppl 4th Report), 555–576 (2004)
W.A. Marshall, J.M. Tanner, Variations in pattern of pubertal changes in girls. Arch. Dis. Child 44, 291–303 (1969)
S. Richards, N. Aziz, S. Bale, D. Bick, S. Das, J. Gastier-Foster et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–24 (2015)
W.L. Miller, Assignment of the gene for adrenal P450c17 (steroid 17 alpha hydroxylase/17,20 lyase) to human chromosome 10. J. Clin. Endocrinol. Metab. 63, 789–791 (1986)
D. Türkkahraman, T. Guran, H. Ivison et al. Identification of a novel large CYP17A1 deletion by MLPA analysis in a family with classic 17a-hydroxylase deficiency. Sex. Dev. 9, 91–97 (2015)
S. Nakajin, J.E. Shively, P.M. Yuan et al. Microsomal cytochrome P-450 from neonatal pig testis: two enzymatic activities (17a-hydroxylase and c17,20-lyase) associated with one protein. Biochemistry 20(14), 4037–4042 (1981)
D. Levran, I. Ben-Shlomo, C. Pariente, J. Dor, S. Mashiach, A. Weissman, Familial partial 17,20-desmolase and 17alpha-hydroxylase deficiency presenting as infertility. J. Assist. Reprod. Genet. 20, 21–28 (2003)
C.A. Marsh, R.J. Auchus, Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17‑hydroxylase/17,20‑lyase deficiency and isolated 17,20‑lyase deficiency. Fertil. Steril. 101, 317–322 (2014)
J. Qiao, B. Han, B.L. Liu, W. Liu, J.J. Wu, C.M. Pan, H. Jiang, T. Gu, B.R. Jiang, H. Zhu, Y.L. Lu, W.L. Wu, M.D. Chen, H.D. Song, A unique exonic splicing mutation in the CYP17A1 gene as the cause for steroid 17{alpha}-hydroxylase deficiency. Eur. J. Endocrinol. 164, 627–633 (2011).
M. Zhang, S. Sun, Y. Liu, H. Zhang, Y. Jiao, W. Wang, X. Li, New, recurrent, and prevalent mutations: Clinical and molecular characterization of 26 Chinese patients with 17alpha hydroxylase/17,20-lyase deficiency. J. Steroid Biochem. Mol. Biol. 150, 11–16 (2015)
F. Yao, S. Huang, X. Kang, W. Zhang, P. Wang, Q. Tian, CYP17A1 mutations identified in 17 Chinese patients with 17α-hydroxylase/17,20-lyase deficiency. Gynecol. Endocrinol. 29, 10–15 (2013).
R.J. Auchus, The genetics, pathophysiology, and management of human deficiencies of P450c17. Endocrinol. Metab. Clin. North Am. 30(1), 101–119 (2001)
W.L. Miller, J.C. Acherman, C.E. Flück. The adrenal cortex and its disorder. Pediatric Endocrinology. 3rd ed. M.A. Sperling editor. Saunders Elsevier, Philadelphia, PA, USA), 2008) 444–511
A. Biason-Lauber, M. Boscaro, F. Mantero, G. Balercia, Defects of steroidogenesis. J. Endocrinol. Investig. 33, 756–766 (2010).
R. Voutilainen, W.L. Miller, Developmental expression of genes for the stereoidogenic enzymes P450scc (20,22-desmolase), P450c17 (17 a-hydroxylase/17,20-lyase), and P450c21 (21-hydroxylase) in the human fetus. J. Clin. Endocrinol. Metab. 63(5), 1145–1150 (1986)
Y.M. Kim, M. Kang, J.H. Choi, B.H. Lee, G.H. Kim, J.H. Ohn, S.Y. Kim, M.S. Park, H.W. Yoo, A review of the literature on common CYP17A1 mutations in adults with 17-hydroxylase/17,20 lyase deficiency, a case series of such mutations among Koreans and functional characteristics of a novel mutation. Metab. Clin. Exp. 63, 42–49 (2014)
C. Scaroni, G. Opocher, F. Mantero, Renin-angiotensin-aldosterone system: a long-term follow-up study in 17 alpha-hydroxylase deficiency syndrome (17OHDS). Clin. Exp. Hypertens. A 8(4-5), 773–80 (1986)
Y.M. Bee, C. Manju, M. Papari-Zareei et al. Phenotypic variation in a Chinese family with 46,XY and 46,XX 17a-hydroxylase deficiency. Gynecol. Endocrinol. 28(4), 322–325 (2012)
Y.P. Wang, Y.J. Zhao, G.Y. Zhou, B. He, CYP17A1 gene mutations and hypertension variations found in 46,XY females with combined 17α-hydroxylase/17, 20-lyase deficiency. Gynecol. Endocrinol. 30, 456–460 (2014).
E.L. Van Den Akker, J.W. Koper, A.L. Boehmer et al. Differential inhibition of 17alpha-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J. Clin. Endocrinol. Metab. 87(12), 5714–5721 (2002)
K. Mussig, S. Kaltenbach, F. Machicao et al. 17alphahydroxylase/17,20- lyase deficiency caused bya novel homozygous mutation (Y27Stop) in the cytochrome CYP17 gene. J. Clin. Endocrinol. Metab. 90, 4362–4365 (2005)
F.E. Herrera, E. Espín, A.M.T. Álvarez, L.J. Beltrán, D.G. Correa, G. Burgos, A. Llamos, C. Zurita, S. Rojas, I.D. Espín, K.C. Ludeña, J.S. Vega, J.P. Basto, A report of congenital adrenal hyperplasia due to 17α-hydroxylase deficiency in two 46,XX sisters. Gynecol. Endocrinol. 36(1), 24–29 (2020)
C.E. Kater, E.G. Biglieri, Disorders of steroid 17a-hydroxylase deficiency. Endocrinol. Metab. Clin. North Am. 23(2), 341–357 (1994)
Author contributions
Surgical and medical practices: A.B., S.B., E.U., A.A.K., M.T., and Y.K.H. Concept: A.B., S.B., E.U., A.A.K., M.T., and Y.K.H. Design: A.B., S.B., E.U., A.A.K., R.E., and Y.K.H. Data collection or processing: A.B., S.B. Analysis or interpretation: A.B., S.B., E.U., R.E., and Y.K.H. Literature search: A.B., E.U., R.E. Writing: A.B., E.U.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors. The study was performed in accordance with the rules of Declaration of Helsinki and approved by the Institutional Ethics Committee of Dicle University Medical School. The study was approved by the Dicle University Faculty of Medicine Clinical Research Ethics Committee (approval number: 2021/165).
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Beştaş, A., Bolu, S., Unal, E. et al. A rare cause of delayed puberty and primary amenorrhea: 17α-hydroxylase enzyme deficiency. Endocrine 75, 927–933 (2022). https://doi.org/10.1007/s12020-021-02914-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-021-02914-8