
Abstract
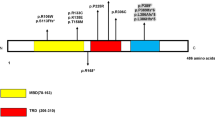
Rett syndrome is a severe neurodevelopmental disorder, almost exclusively affecting females and characterized by a wide spectrum of clinical manifestations. Both the classic and atypical forms of Rett syndrome are primarily due to mutations in the methyl-CpG-binding protein 2 (MECP2) gene. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene have been identified in patients with atypical Rett syndrome, X-linked infantile spasms sharing common features of generally early-onset seizures and mental retardation. CDKL5 is known as serine/threonine protein kinase 9 (STK9) and is mapped to the Xp22 region. It has a conserved serine/threonine kinase domain within its amino terminus and a large C-terminal region. Disease-causing mutations are distributed in both the amino terminal domain and in the large C-terminal domain. We have screened the CDKL5 gene in 44 patients with atypical Rett syndrome who had tested negative for MECP2 gene mutations and have identified 6 sequence variants, out of which three were novel and three known mutations. Two of these novel mutations p.V966I and p.A1011V were missense and p.H589H a silent mutation. Other known mutations identified were p.V999M, p.Q791P and p.T734A. Sequence homology for all the mutations revealed that the two mutations (p.Q791P and p.T734A) were conserved across species. This indicated the importance of these residues in structure and function of the protein. The damaging effects of these mutations were analysed in silico using PolyPhen-2 online software. The PolyPhen-2 scores of p.Q791P and p.T734A were 0.998 and 0.48, revealing that these mutations could be deleterious and might have potential functional effect. All other mutations had a low score suggesting that they might not alter the activity of CDKL5. We have also analysed the position of the mutations in the CDKL5 protein and found that all the mutations were present in the C-terminal domain of the protein. The C-terminal domain is required for cellular localization through protein–protein interaction; any mutations in this domain might alter this function of the protein. This is the first report from India showing the mutation in CDKL5 gene in Indian cases of Rett syndrome. Our study emphasizes the role of CDKL5 mutation screening in cases of atypical Rett syndrome with congenital seizure variant.




Similar content being viewed by others


References
Allen, R. C., Zoghbi, H. Y., Moseley, A. B., Rosenblatt, H. M., & Belmont, J. W. (1992). Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. American Journal of Human Genetics, 51(6), 1229–1239.
Amir, R. D., Van den Veyver, I. B., Wan, M., Tra, C. Q., Francke, U., & Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MeCP2, encoding methyl CpG binding protein 2. Nature Genetics, 23(2), 185–188.
Archer, H. L., Evans, J., Edwards, S., Colley, J., Newbury-Ecob, R., O’Callaghan, F., et al. (2006). CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. Journal of Medical Genetics, 43(9), 729–734.
Bahi-Buisson, N., & Bienvenu, T. (2012). CDKL5-related disorders: From clinical description to molecular genetics. Molecular Syndromology, 2(3–5), 137–152.
Bahi-Buisson, N., Villeneuve, N., Caietta, E., Jacquette, A., Maurey, H., Matthijs, G., et al. (2012). Recurrent mutations in the CDKL5 gene: Genotype-phenotype relationships. American Journal of Medical Genetics Part A, 158A(7), 1612–1619.
Bertani, I., Rusconi, L., Bolognese, F., Forlani, G., Conca, B., De Monte, L., et al. (2006). Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. Journal of Biological Chemistry, 281(42), 32048–32056.
Castren, M., Gaily, E., Tengström, C., Lahdetie, J., Archer, H., & Ala-Mello, S. (2010). Epilepsy caused by CDKL5 mutations. European Journal of Paediatric Neurology, 15(1), 65–69.
Cheadle, J. P., Gill, H., Fleming, N., Maynard, J., Kerr, A., Leonard, H., et al. (2000). Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: Correlation of disease severity with mutation type and location. Human Molecular Genetics, 9(7), 1119–1129.
Evans, J. C., Archer, H. L., Colley, J. P., Ravn, K., Nielsen, J. B., Kerr, A., et al. (2005). Early onset seizures and Rett-like features associated with mutations in CDKL5. European Journal of Human Genetics, 13(10), 1113–1120.
Hadzsiev, K., Polgar, N., Bene, J., Komlosi, K., Karteszi, J., Hollody, K., et al. (2011). Analysis of Hungarian patients with Rett syndrome phenotype for MECP2, CDKL5 and FOXG1 gene mutations. Journal of Human Genetics, 56(3), 183–187.
Hagberg, B. A., & Skjeldal, O. H. (1994). Rett variants: A suggested model for inclusion criteria. Pediatric Neurology, 11(1), 5–11.
Hanefeld, F. (1985). The clinical pattern of the Rett syndrome. Brain Development, 7(3), 320–325.
Jayaram, B., Dhingra, P., Lakhani, B., & Shekhar, S. (2012). Bhageerath—Targeting the near impossible: Pushing the frontiers of atomic models for protein tertiary structure prediction. Journal of Chemical Sciences, 124(1), 83–91.
Mari, F., Azimonti, S., Bertani, I., Bolognese, F., Colombo, E., Caselli, R., et al. (2005). CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Human Molecular Genetics, 14(14), 1935–1946.
Montini, E., Andolfi, G., Caruso, A., Buchner, G., Walpole, S. M., Mariani, M., et al. (1998). Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics, 51(3), 427–433.
Rosas-Vargas, H., Bahi-Buisson, N., Philippe, C., Nectoux, J., Girard, B., N’Guyen Morel, M. A., et al. (2008). Impairment of CDKL5 nuclear localisation as a cause for severe infantile encephalopathy. Journal of Medical Genetics, 45(3), 172–178.
Roy, A., Kucukural, A., & Zhang, Y. (2010). I-TASSER: A unified platform for automated protein structure and function prediction. Nature Protocol, 5(4), 725–738.
Rusconi, L., Salvatoni, L., Giudici, L., Bertani, I., Kilstrup-Nielsen, C., Broccoli, V., et al. (2008). CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. Journal of Biological Chemistry, 283(44), 30101–30111.
Scala, E., Ariani, F., Mari, F., Caselli, R., Pescucci, C., Longo, I., et al. (2005). CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. Journal of Medical Genetics, 42(2), 103–107.
Tao, J., Van Esch, H., Hagedorn-Greiwe, M., Hoffmann, K., Moser, B., Raynaud, M., et al. (2004). Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. American Journal of Human Genetics, 75(6), 1149–1154.
Van Esch, H., Jansen, A., Bauters, M., Froyen, G., & Fryns, J. P. (2007). Encephalopathy and bilateral cataract in a boy with an interstitial deletion of Xp22 comprising the CDKL5 and NHS genes. American Journal of Medical Genetics Part A, 143(4), 364–369.
Villard, L., Kpebe, A., Cardoso, C., Chelly, B., Tardieu, P., & Fontes, M. (2000). Two affected boys in a Rett syndrome family. Neurology, 55(8), 1188–1193.
Weaving, L. S., Christodoulou, J., Williamson, S. L., Friend, K. L., McKenzie, O. L. D., Archer, H., et al. (2004). Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. American Journal of Human Genetics, 75(6), 1079–1093.
Zoghbi, H. Y., Percy, A. K., Schultz, R. J., & Fill, C. (1990). Patterns of X chromosome inactivation in the Rett syndrome. Brain Development, 12(1), 131–135.
Acknowledgments
We thank all the patients and their families for contribution to this study. The authors are also thankful to Director, NIRRH, for providing necessary facilities and Indian Council of Medical Research (ICMR) and Department of Science & Technology (SR/FT/LS-87/2010), Govt of India, for providing financial grant for the study. The help in computational analysis provided by Dr Susan Thomas; Saravanan & Nanda in sequencing are also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig 1
Prediction of 3-dimensional CDKL5 protein structures using online platform (a) using I-Tasser (b) using Bhageerath H (TIFF 25503 kb)
Rights and permissions
About this article
Cite this article
Das, D.K., Mehta, B., Menon, S.R. et al. Novel Mutations in Cyclin-Dependent Kinase-Like 5 (CDKL5) Gene in Indian Cases of Rett Syndrome. Neuromol Med 15, 218–225 (2013). https://doi.org/10.1007/s12017-012-8212-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-012-8212-z