
Abstract
As the diagnosis of systemic sclerosis (SSc) is generally suggested by the presence of Raynaud’s phenomenon followed by typical skin thickening associated with the presence of additional extracutaneous features, capillaroscopic abnormalities, and characteristic autoantibodies, the first classification criteria, published by the American Rheumatism Association in 1980, were based only on clinical and chest X-ray items. As a consequence, 10% to 20% of the patients did not meet these criteria. In 1988, an international consensus was reached resulting in the proposal of a new and more practical classification based on the judgment and clinical practice of an expert panel. This classification introduced the SSc nail fold capillaroscopy abnormalities (dilation and/or avascular areas) and specific antinuclear antibodies. Two subsets of SSc emerged from discussions: diffuse cutaneous SSc (dcSSc) and limited cutaneous SSc (lcSSc). The calcifications, Raynaud’s phenomenon, esophageal hypomotility, sclerodactyly, and telangiectasia (CREST) syndrome can be considered an lcSSc. In 2001, LeRoy and Medsger, realizing the shortcomings of the 1988 subsets in being too exclusive and taking advantage of increased experience with nail fold capillaroscopy and autoantibody determination, proposed criteria for an additional early or limited subset of SSc (lSSc), to supplement the previously recognized lcSSc and dcSSc forms. Patients with lSSc must have Raynaud’s phenomenon and SSc-specific nail fold capillary changes and/or SSc-specific autoantibodies. Some lSSc patients who have no cutaneous involvement but common SSc nail fold capillaroscopy abnormalities, specific antinuclear antibodies, and visceral involvement are sometimes called SSc sine scleroderma. Whether or not lSSc and SSc sine scleroderma are the same or two different subsets is currently not known.
Similar content being viewed by others


Systemic sclerosis (SSc) is a connective tissue disease of unknown origin characterized by fibrosis of the skin (a hallmark of the disease) and internal organs, associated with severe microangiopathy and T lymphocyte activation with autoantibodies and a possible role of B cells in fibrosis. SSc preferentially affects women (female-to-male ratio approximately 8:2) and generally begins between 45 and 65 years. This connective tissue disease has a worldwide distribution. The exact prevalence remains unclear with a considerable disparity between areas and countries: reports range from 30.8 to 286 cases per million. A recent study of SSc in the Detroit area (USA) based on a capture–recapture analysis estimated the prevalence to be 276 cases per million adults [1]. Using the same methodology, Le Guern et al. reported the prevalence of SSc in Seine-Saint-Denis (a suburb of Paris) to be 158.3 (95% confidence interval, 129–187) per million adults [2]. The 10-year survival rate varies from 60% to 90% depending on the cutaneous extension [3]. Survival also depends on whether or not there is visceral involvement, interstitial lung disease and pulmonary arterial hypertension currently being the two major causes of death [4].
Diagnosis
The diagnosis of SSc is generally suggested by the presence of typical skin thickening and hardening (sclerosis) which usually begins with the fingers. Raynaud’s phenomenon usually precedes the skin involvement by several weeks to several years. The diagnosis is supported by the presence of additional extracutaneous features, capillaroscopic abnormalities, and characteristic autoantibodies. A skin biopsy is not usually required in order to confirm the diagnosis but may sometimes be needed to differentiate SSc from other syndromes such as eosinophilic fasciitis, scleredema, or scleromyxedema.
Patients generally have variable degrees of skin sclerosis. The greatest degree of skin involvement is observed on the fingers, hands, and face, especially in the early stages of the disease. The diagnosis may be difficult during the first few months of the disease. At this early stage of the disease, soft tissue swelling rather than skin induration may be the most prominent feature (puffy fingers). Other cutaneous features, such as calcinosis or telangiectasia, may be helpful in confirming the diagnosis but are often absent in the early stages.
The combination of skin induration and one or more of the following clinical features supports the diagnosis of SSc:
-
Heartburn and/or dysphagia of new onset due to distal esophageal dysmotility
-
Acute onset of hypertension and renal insufficiency
-
Dyspnea on exertion associated with interstitial lung disease
-
Dyspnea on exertion associated with pulmonary arterial hypertension
-
Diarrhea with malabsorption or intestinal pseudo-obstruction
-
Facial, tongue, lip, or hand telangiectasia
-
Digital ulcer and or digital pitting scar
-
Typical microvascular changes on nail fold capillaroscopy.
The presence of characteristic autoantibodies is supportive of the diagnosis of SSc. Antinuclear antibodies are present in up to 90% of SSc patients [5]. Specific autoantibodies include anticentromere (ACA), antitopoisomerase-I (Scl-70), anti-RNA polymerase, or U3-RNP antibodies. Antinuclear antibody positivity (with a nucleolar staining pattern) is also frequently observed. However, the diagnosis of SSc is not excluded by the absence of antinuclear antibodies [6].
Some SSc patients also have signs of other defined connective tissue diseases, such as systemic lupus erythematosus, rheumatoid arthritis, and polymyositis. This is called overlap syndrome and occurs in roughly 10% of SSc patients [7].
Classification
Classification criteria are an essential component of SSc research as they ensure that patients recruited for studies have similar features and allow results to be compared across studies. There has been increasing recognition of subsets within the spectrum of SSc, in the belief that subsets of patients differ in terms of disease expression, response to therapy, morbidity, and prognosis. Thus, the accurate identification of disease subsets may improve the ability to prognosticate organ involvement and survival, develop appropriate screening programs, and guide treatment recommendations [8].
The 1980 ARA criteria for SSc
The first aim of the 1980 American Rheumatism Association (ARA) classification criteria for SSc [9] (Table 1) was to distinguish SSc from non-SSc. SSc cases and non-SSc cases were submitted by 29 centers based on their own expertise. After Committee review, 797 cases were included in the study for analysis. There were 264 definite SSc cases, 35 probable or early stage SSc cases, 85 overlap syndrome cases, 172 systemic lupus erythematosus cases, 120 polymyositis/dermatomyositis cases, and 121 cases of Raynaud’s phenomenon not caused by the previous disorders. Among the 264 definite SSc cases and 35 probable SSc cases, 91% and 51%, respectively, had proximal skin involvement. For this reason, proximal scleroderma was defined as a major criterion with a high sensitivity and specificity for the diagnosis (91% and 99%, respectively). It is important to point out that this classification was developed using data from patients with a connective tissue disease, and thus, while it makes it possible to discriminate between SSc patients and patients with another connective tissue disease, it is not intended for use in the diagnosis of SSc beyond this context. Moreover, the 1980 ARA criteria make no attempt to deal with disease heterogeneity. This first classification suggested two major subsets of SSc, the first one with proximal cutaneous sclerosis (diffuse form) and the second one without proximal cutaneous sclerosis (i.e., a more limited form).
To evaluate these proposed criteria, the Pittsburgh cohort (1972–1983) of 639 patients with SSc, comprising 315 with diffuse SSc (49%) and 324 with limited SSc, was tested [10]. Of the latter group, 134 (21% of total SSc, 41% of limited SSc) did not fulfill the major preliminary 1980 ARA criterion, and 67 (10.5% of total SSc, 20% of limited SSc) did not fulfill either the major or two of the three minor criteria [10]. In a second publication, Lonzetti et al. reviewed the records of 259 French Canadian patients, diagnosed as definite SSc, 29 cases of which were considered diffuse (truncal skin involvement), 78 intermediate (skin involvement not truncal, but proximal to metacarpophalangeal joints), and 152 limited (sclerodactyly only, plus Raynaud’s phenomenon) [11]. Two thirds of the 152 patients with limited SSc did not fulfill the 1980 ARA preliminary criteria. Nonetheless, 50% of the limited SSc patients were ACA antibody positive, and two thirds had significant nail fold capillary abnormalities. By adding nail fold capillary findings and ACA, the sensitivity of the 1980 ARA criteria improved from 33.4% to 91.5%. In a more recent publication comparing 87 SSc patients from France and 240 SSc patients from the USA [12], 76/87 (13%) of French patients and 199/240 (17%) of the American patients did not meet the 1980 ARA criteria for SSc. This underlines the need for a more precise classification.
Limited versus diffuse cutaneous SSc: the 1988 LeRoy et al. classification
In 1988, an international consensus was reached resulting in the proposal of a new and more practical classification based on the judgment and clinical practice of an expert panel [13]. Interesting is this classification introduced SSc nail fold capillaroscopy abnormalities and specific antinuclear antibodies. Two subsets of SSc emerged from discussions: diffuse cutaneous SSc (dcSSc) and limited cutaneous SSc (lcSSc) [13]. When skin thickening is distal to the metacarpophalangeal joints, it is called sclerodactyly. The question of whether dcSSc and lcSSc are different diseases or represent different phenotypes of the same disease has not yet been settled, but transition from one to the other is seldom seen, which could favor the former interpretation (Table 2). Importantly, the two classical specific SSc autoantibodies segregate clearly between the subsets, lcSSc being associated with ACA and dcSSc with antitopoisomerase antibodies. If this classification were applied to the 639 Pittsburgh cohort patients [10], most of these SSc patients would be expected to fall within one or other of these SSc subsets. This subdivision may identify pathogenetically distinct subsets. Patients with dcSSc are more prone than lcSSc patients to develop severe lung, renal, or myocardial involvement, particularly during the first 3 to 5 years of SSc evolution [14] and more frequently have antitopoisomerase autoantibodies. For example, the risk of developing a scleroderma renal crisis is higher in dcSSc than in lcSSc patients, with an odds ratio above 7 compared with [15]. Consequently, the prognosis and survival from onset of the disease is worse in dcSSc than in lcSSc. The lcSSc/dcSSc classification has been widely accepted and used in numerous clinical studies and therapeutic trials.
Calcifications, Raynaud’s phenomenon, esophageal hypomotility, sclerodactyly, and telangiectasia (CREST) syndrome can be considered as a lcSSc. This subset was first described as a benign form of SSc [16]. CREST patients could have three, four, or all five of the syndrome features, and the components were probably never meant to serve as criteria for a special subset of SSc patients. Features of CREST occur with time irrespective of other disease characteristics. Moreover, calcifications, Raynaud’s phenomenon, esophageal hypomotility, and telangiectasia are also common in dcSSc [17]. Therefore, the use of CREST to define a subset of SSc patients is not helpful and may in fact be confusing albeit some lcSSc patients sometimes have a particular presentation with many calcifications and lots of telangiectasia.
Limited SSc: the 2001 LeRoy et al. classification
In 2001, LeRoy and Medsger, realizing the shortcomings of the 1988 subsets in being too exclusive and taking advantage of the increased experience with nail fold capillaroscopy and autoantibody determination, proposed criteria for an additional early or limited subset of SSc (lSSc), to supplement the previously recognized lcSSc and dcSSc forms (Table 3) [18]. Patients with lSSc must have Raynaud’s phenomenon and SSc-specific nail fold capillary changes and/or SSc-specific autoantibodies. Some SSc patients, early in their disease course, may present to clinic without skin involvement, but the SSc diagnosis should be based on clinical and serologic findings. Patients with lSSc may or may not later develop lcSSc or dcSSc. It is not yet clear whether lSSc should be considered an early stage of the disease. However, it is important to include such patients in the prospective population database since they may be informative regarding the development of typical SSc organ involvement. Nevertheless, the proposal for lSSc has not gained wide acceptance, perhaps because lSSc and lcSSc are easily confused.
SSc sine scleroderma
Some lSSc patients who have no cutaneous involvement but have SSc nail fold capillaroscopy abnormalities, specific antinuclear antibodies, and visceral involvement are sometimes called SSc sine scleroderma [19]. Whether or not lSSc and SSc sine scleroderma are the same or two different subsets is currently not known. Using the Pittsburgh scleroderma databank, Poormoghim et al. [19] identified 66 SSc patients with no skin changes, who were called SSc sine scleroderma. Eighteen of the patients who initially had no skin changes later developed sclerodactyly and were classified as having lcSSc. The mean duration from the first Pittsburgh visit to the development of sclerodactyly was 3.9 years (median 3.6 years). These 18 lcSSc patients were added to a previously identified 490 lcSSc comparative group. Thus, the final study group consisted of 48 patients with no skin thickening at the time of first evaluation or at any time during follow-up (SSc sine scleroderma group) and 507 patients with lcSSc. The mean duration from symptom onset to study entry was 15.1 years for SSc sine scleroderma patients and 20.7 years for lcSSc patients. At the time of their final evaluation, the 48 SSc sine scleroderma patients had a mean total disease duration of 18.6 years since onset and had not developed lcSSc or any other connective tissue disease. When the SSc sine scleroderma patients were stratified according to age, sex, and disease duration from onset, there were no differences in the frequencies of symptoms and organ involvement. Bibasal fibrosis on chest X-ray concerned 39% of SSc sine scleroderma patients compared to 37% of lcSSc patients and pulmonary arterial hypertension 23% versus 13%, respectively (p = NS). Not all patients were tested for antinuclear antibodies, but there were no significant differences in the frequencies with which ACA were detected in the two groups (31% in SSc sine scleroderma and 54% in the lcSSc, p < 0.09). Thus, it is difficult to consider SSc sine scleroderma as an early stage of the disease. We can probably consider lSSc as sometimes representing an early stage of the disease if symptoms have evolved for less than 5 years, but clearly, after 5 years’ evolution, especially if organ involvement occurs, these patients should be considered as having another subset of SSc and should be called SSc sine scleroderma. We have already seen patients of this type who never go on to develop skin thickening.
Like Poormoghim et al. [19], we believe that a patient should be considered to have SSc sine scleroderma if he or she has all of the following features:
-
1.
Raynaud’s phenomenon or a peripheral vascular equivalent (digital pitting scars, digital-tip ulcers, digital-tip gangrene, and abnormal nail fold capillaries);
-
2.
positive antinuclear antibodies;
-
3.
any one of the following: distal esophageal hypomotility, small bowel hypomotility, pulmonary interstitial fibrosis, primary pulmonary arterial hypertension (without fibrosis), cardiac involvement typical of scleroderma, or renal failure consistent with a scleroderma renal crisis; and
-
4.
no other defined connective tissue or other disease as a cause of (1), (2), or (3).
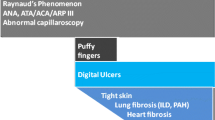
Besides the 1980 ARA criteria for SSc [9], the 1988 LeRoy et al. classification [13] gives us the opportunity to distinguish the two most frequent subsets of SSc: lcSSc and dcSSc, which clearly correspond to two distinct clinical, biological, and prognosis subsets. In an individual patient with lcSSc, the appearance of truncal or proximal limbs skin involvement clearly changes the patient from the lcSSc subset to the dcSSc subset (this is possible during the first 3 to 5 years of SSc evolution). Nevertheless, the regression of truncal or proximal limb skin involvement to normal after several years in a patient with dcSSc does not warrant a change from dcSSc to lcSSc. The 2001 LeRoy et al. classification [18] gives us the opportunity to identify a new subset called lSSc, without cutaneous involvement, with the help of capillaroscopy and antinuclear antibodies. Given the likelihood that lSSc sometimes correspond to an early stage of SSc that may or may not evolve to an lcSSc or dcSSc subset, SSc sine scleroderma probably corresponds to a subset of patients who initially have an lSSc and progressively develop organ involvement over time but never have skin thickening (Fig. 1). The major problem, in case of lSSc associating Raynaud’s phenomenon and abnormal widefield nail fold capillaroscopy and/or SSc selective autoantibodies only, is to give a patient who will possibly never develop any organ involvement the diagnosis of SSc, which carries with it the risk of an adverse psychological effect. The European League Against Rheumatism (EULAR) has created the EULAR Scleroderma Trials and Research group (EUSTAR) in order to coordinate all European centers to fight the disease (www.eustar.org). This network is the platform from which a better understanding of the disease in its earliest phase will appear.

The four possible evolutions of limited systemic sclerosis
References
Mayes MD, Lacey JV Jr, Beebe-Dimmer J et al (2003) Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 48:2246–2255
Le Guern V, Mahr A, Mouthon L, Jeanneret D, Carzon M, Guillevin L (2004) Prevalence of systemic sclerosis in a French multi-ethnic county. Rheumatology (Oxford) 43:1129–1137
Scussel-Lonzetti L, Joyal F, Raynauld JP et al (2002) Predicting mortality in systemic sclerosis: analysis of a cohort of 309 off French Canadian patients with emphasis on features at diagnosis as predictive factors for survival. Medicine (Baltimore) 81:154–167
Steen VD, Medsger TA (2007) Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis 66:940–944
Vonk MC, Broers B, Heijdra YF et al (2008) Systemic sclerosis and its pulmonary complications in The Netherlands: an epidemiological study. Ann Rheum Dis 68:961–965
Reveille JD, Solomon DH, American College of Rheumatology Ad Hoc Committee of Immunologic Testing Guidelines (2003) Evidence-based guidelines for the use of immunologic tests: anticentromere, Scl-70, and nucleolar antibodies. Arthritis Rheum 49:399–412
Hunzelmann N, Genth E, Krieg T et al (2008) The registry of the German Network for Systemic Scleroderma: frequency of disease subsets and patterns of organ involvement. Rheumatology 47:1185–1192
Johnson SR, Feldman BM, Hawker GA (2007) Classification criteria for systemic sclerosis subsets. J Rheumatol 34:1855–1863
Masy AT, Rodnan GP, Medsger TA Jr, Altman RD, D’angelo WA, Fries JF, the Subcommittee for Scleroderma Criteria of American Rheumatism Association Diagnostic and Therapeutic Criteria Committee (1980) Preliminary criteria for the classification of systemic sclerosis (scleroderma). Arthritis Rheum 23:581–590
Medsger TA Jr (1985) Comment on Scleroderma Criteria Cooperative Study. In: Black CM, Myers AR (eds) Current topics in rheumatology: systemic sclerosis (scleroderma). Gower Medical Publishing, New York, pp 16–17
Lonzetti LS, Joyal F, Raynauld JP et al (2001) Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 44:735–736
Meyer OC, Fertig N, Lucas M, Somogyi N, Medsger TA Jr (2007) Disease subsets, antinuclear antibody profile, and clinical features in 127 French and 247 US adult patients with systemic sclerosis. J Rheumatol 34:104–109
LeRoy EC, Black C, Fleischmajer R et al (1988) Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 15:202–205
Medsger TA Jr (2003) Natural history of systemic sclerosis and assessment of disease activity, severity, functional status, and psychologic well-being. Rhum dis Clin N Am 29:255–273
Penn H, Howie AJ, Kingdon EJ, Bunn CCRJ et al (2007) Scleroderma renal crisis: patient characteristics and long-term outcomes. Q J Med 100:485–494
Winterbauer RH (1964) Multiple telangiectasia, Raynaud’s phenomenon, sclerodactyly, and subcutaneous calcinosis: a syndrome mimicking hereditary hemorrhagic telangiectasia. Bull Johns Hopkins Hosp 114:361–383
Barnett AJ, Miller M, Littlejohn GO (1988) The diagnosis and classification of scleroderma (systemic sclerosis). Postgrad Med J 64:121–125
LeRoy EC, Medsger TA Jr (2001) Criteria for the classification of early systemic sclerosis. J Rheumatol 28:1573–1576
Poormoghim H, Lucas M, Fertig N, Medsger TA Jr (2000) Systemic sclerosis sine scleroderma. Demographic, clinical, and serological features and survival in forty-eight patients. Arthritis Rheum 43:444–451
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hachulla, E., Launay, D. Diagnosis and Classification of Systemic Sclerosis. Clinic Rev Allerg Immunol 40, 78–83 (2011). https://doi.org/10.1007/s12016-010-8198-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-010-8198-y