
Abstract
Directed differentiation of adult multipotent stromal cells (MSC) is critical for effective treatment strategies. This study was designed to evaluate the capability of equine MSC from bone marrow (BMSC) and adipose tissue (ASC) on a type I collagen (COLI) scaffold to undergo chondrogenic, osteogenic and adipogenic differentiation and form extracellular matrix (ECM) in vitro. Following determination of surface antigen expression, MSC were loaded into scaffolds in a perfusion bioreactor and loading efficiency was quantified. Cell-scaffold constructs were assessed after loading and 7, 14 and 21 days of culture in stromal or induction medium. Cell number was determined with DNA content, cell viability and spatial uniformity with confocal laser microscopy and cell phenotype and matrix production with light and scanning electron microscopy and mRNA levels. The MSC were positive for CD29 (>90 %), CD44 (>99 %), and CD105 (>60 %). Loading efficiencies were >70 %. The ASC and BMSC cell numbers on scaffolds were affected by culture in induction medium differently. Viable cells remained uniformly distributed in scaffolds for up to 21 days and could be directed to differentiate or to maintain an MSC phenotype. Micro- and ultrastructure showed lineage-specific cell and ECM changes. Lineage-specific mRNA levels differed between ASC and BMSC with induction and changed with time. Based on these results, equine ASC and BMSC differentiate into chondrogenic, osteogenic and adipogenic lineages and form ECM similarly on COLI scaffolds. The collected data supports the potential for equine MSC-COLI constructs to support diverse equine tissue formation for controlled biological studies.
Similar content being viewed by others
Introduction
Multipotent stromal cells (MSC) in combination with biocompatible scaffolds may augment current therapies to restore equine tissue damaged by trauma or disease [1, 2]. In vitro expansion and mesenchymal lineage differentiation efficiencies are not identical between adult equine bone marrow- (BMSC) and adipose (ASC) derived multipotent stromal cells, two of the best characterized equine MSC [3–6]. While outcomes vary among species and culture conditions, it is often thought that BMSC have better chondrogenic and osteogenic capabilities while ASC seem to have the best adipogenesis [4, 5]. The tissue of origin may influence the ability of MSC to differentiate along specific lineages [7]. Hence, cells from different tissues may demonstrate varying differentiation capacities under identical culture conditions.
Biocompatible scaffolds that replicate target tissue microenvironments effectively support tissue formation by MSC [8]. Collagen is a major component of ECM, and the cell interactions with collagen matrix are thought to closely resemble those in the native cell-tissue milieu [9]. Scaffolds composed of collagen are bioabsorbable, have high cell affinity and support multipotent cell proliferation [10–13]. Collagen based scaffolds in combination with undifferentiated cells support diverse tissue generation including adipose [13], bone [14], cartilage [15, 16], bladder [17], heart [18] and skin [19], among others. Type I collagen (COLI) implants are commercially available [20]. Comprehensive in vitro evaluations of interactions between COLI scaffold and equine ASC and BMSC, are necessary to establish the potential of equine MSC-COLI constructs to support diverse mesenchymal tissue generation. The resulting information is critical for future generation of custom tissue implants to augment fracture, wound and intra-articular therapies.
Bioreactors provide controlled environments for cell loading and significantly contribute to the density and uniformity of cell distribution [21–23]. Compared to static methods of cell loading like droplet and cell suspension, dynamic cell loading results in higher seeding efficiency and more even cell distribution [23]. Additionally, cells seeded by perfusion tend to have more uniform distribution compared to static and spinner flask methods of cell loading [22]. High cell loading efficiency reduces the initial cell numbers required to reach optimum scaffold cell densities and thus decreases cell expansion time [24], and uniform cell loading throughout the scaffold contributes to homogenous tissue generation [25]. Use of a perfusion bioreactor to load adult equine MSC onto biocompatible scaffolds may therefore positively impact in vitro generation of tissue-specific equine MSC-scaffold constructs designed to direct tissue formation following implantation.
A series of related experiments were conducted to characterize adult equine BMSC and ASC mesenchymal trilineage differentiation and extracellular matrix production on COLI scaffolds. The first hypothesis tested was that equine BMSC and ASC have indistinguishable cell loading efficiency, viability, and spatial distribution on COLI scaffolds under identical perfusion bioreactor loading conditions and subsequent culture. The second hypothesis was that under matching adipogenic, osteogenic, and chondrogenic culture conditions, BMSC on COLI scaffolds have higher target tissue messenger ribonucleic acid (mRNA) levels and support more efficient osteogenic and chondrogenic extracellular matrix (ECM) formation than ASC on COLI scaffolds, which have superior outcomes for adipogenesis.
Materials and Methods
Study Design
Tissue harvest procedures were approved by the Institutional Animal Care and Use Committee (protocol #11-047). Stromal cells from four thoroughbred geldings (7–10 years) were isolated immediately after tissue harvest from sternal bone marrow and subcutaneous adipose over the gluteal musculature [4, 5]. Cryopreserved cell passage (P) 1 adult equine ASC and BMSC were revitalized and expanded to P3. For purposes of this study, all expansion procedures were performed in T75 flasks with a seeding density of 5 × 103 cells/cm2 and cell passage at 75 % confluence. Cells (106 cells/scaffold) were loaded into bovine corium COLI (AviteneTM UltrafoamTM Collagen Sponge, Davol Inc., subsidiary of C.R. Bard, Inc. Warwick, RI, USA) in perfusion bioreactors. Loading efficiency was evaluated immediately after cell seeding. Constructs were cultured in stromal (basal), adipogenic, osteogenic or chondrogenic medium. Viability, spatial distribution on COLI scaffolds and construct microstructure were determined with confocal laser scanning and light microscopy, respectively, immediately after loading and 7, 14 and 21 days of culture. Deoxyribonucleic acid (DNA) and total ribonucleic acid (RNA) were isolated to quantify cell numbers and target gene mRNA levels at the same time points. Ultrastructure was assessed with scanning electron microscopy (SEM) after 21 days. A total of 12 scaffolds measuring 10 × 7 × 2 mm were prepared from each cell type from individual horses. Following cell loading, scaffolds were divided in half and randomly assigned to a culture medium and time point. There were four replicates for each outcome assessment, which were performed in duplicate for all horses.
Cell Harvest, Expansion and Preservation
Inclusion criteria for cell donors were designed to identify a clinically relevant population with limited variability attributable to breed, age, gender or comorbidities. Inclusion criteria were: 1) thoroughbred; 2) 7–10 years; 3) gelding; 4) normal physical exam. For bone marrow harvest, horses were sedated with detomidine HCl (0.04 mg/kg intravenously (IV)), an area over the cranial sternebrae was aseptically prepared, and surface tissues were desensitized with 2 % lidocaine. Bone marrow aspirates were collected through 10G 3-in bone marrow biopsy needles into heparinized syringes (1,000 IU/10 mL aspirate). The aspirate was diluted 1:1 with stromal medium (Dulbecco’s modified Eagle’s medium (DMEM)-Ham’s F12 (Hyclone, Logan, UT, USA), 1 % antibiotic/antimycotic solution (MP Biomedical, Irvine, CA, USA), 10 % fetal bovine serum (Hyclone)), layered over Ficoll-Paque® PLUS (Stem Cell Technologies, Vancouver, Canada), and centrifuged at 1,400 r.p.m. (350 g) for 30 min at 4 °C. The cell layer was aspirated and centrifuged (1,200 r.p.m. (260 g), 5 min, 4 °C). The cell pellet was then resuspended in 10 mL of stromal medium. The total viable nucleated cell number was determined based on trypan blue exclusion.
Under the same sedation and following aseptic preparation, skin was desensitized over the gluteal musculature with 2 % lidocaine. A 10 cm horizontal skin incision was made with a #10 scalpel blade, and subcutaneous adipose tissue (15 mL) was sharply excised. Care was taken to avoid fascia tissue during the harvest procedure. Skin was opposed with #0 nylon skin sutures in a simple interrupted pattern. Adipose tissue was minced and mixed with an equal volume of phosphate buffered saline (PBS). The mixture was allowed to separate into two phases, and the infranatant was digested for 2 h at 37 °C in an equal volume of PBS with 1 % bovine serum albumin (BSA) and 0.1 % type I collagenase (Worthington Biochemical, Lakewood, NJ, USA). After addition of 5 mL 1 % BSA, the mixture was centrifuged (1,200 r.p.m. (260 g), 5 min, 4 °C), and the resulting stromal vascular fraction (SVF) pellet was resuspended in stromal medium. Viable cell number was determined as above.
Cell isolates were cultured (37 ºC, 5 % CO2) in stromal medium. For purposes of this study, the initial cell isolate is considered the stromal vascular fraction (SVF). The first cell passage is considered P0. At 75 % confluence, SVF cells were detached with 0.05 % trypsin and seeded at 5 × 103 cells/cm2. When the cells (P0) reached 75 % confluence, they were detached as before and cell aliquots (P1, 1 × 106 cells/ml) in cryopreservation medium (80 % fetal bovine serum (FBS), 10 % DMEM, 10 % dimethylsulfoxide) were maintained in liquid nitrogen for a minimum of 30 days.
Immunophenotype–Flow Cytometry
Following thawing (1 min, 37 °C), P1 MSC aliquots from each horse were cultured in stromal medium for 4 days. Cells were passaged at 75 % confluence and seeded at 5 × 103 cells/cm2 (P2). When cells reached 75 % confluence, they were detached with 0.05 % trypsin and aliquots (P3, 5 × 103 cells/horse, n = 4) of each cell type were combined (2 × 105 cells total) in 200 ml PBS and incubated with 5 μg of unlabeled CD29 (α-human, catalog #610468, BD Biosciences, San Jose, CA, USA), unlabeled CD44 (α-feline, equine reactivity, catalogue # CT-BOV2037, Washington State University Monoclonal Antibody Center, Pullman, WA) or CD105-PE (α-human, catalog #12-1057, eBioscience, San Diego, CA, USA) antibodies for 30 min at room temperature. For CD29 and CD44, indirect immunofluorescence was performed by incubation with goat anti-mouse Ig-FITC (Sigma Aldrich, Saint Louis, MO, USA) for 30 min at room temperature. Following incubation, cells were washed with PBS and fixed with 2 % paraformaldehyde. Surface antigen expression was determined based on comparisons with negative controls illustrated on 10,000 even histogram plots using a fluorescence-activated cell sorting flow cytometer (FACSCalibur, BD Biosciences) and software (CellQuest Pro, BD Biosciences). The specificities of antibodies that were not specifically designated as equine reactive were confirmed by standard western blot band colocalization with human MSC controls at the level provided by the manufacturer [26].
Collagen Scaffold
The scaffold material used in this study was composed of a commercially available collagen sponge (AviteneTM UltrafoamTM Collagen Sponge, Davol Inc., subsidiary of C.R. Bard, Inc. Warwick, RI, USA). The purified bovine corium collagen sponge is porous, pliable, water insoluble and bioabsorbable. Sterile 8 × 12.5 × 0.3 cm sheets were cut into 14 × 10 × 3 mm pieces under aseptic conditions. Saturation of the scaffold material with medium resulted in 30 % shrinkage to give final dimensions of 10 × 7 × 2 mm. Hydrated, individual scaffolds were loaded with cells within perfusion bioreactors as described below.
Loading Efficiency
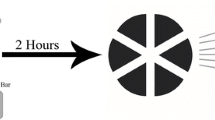
Following revitalization and culture expansion of P1 cell aliquots as described above, cells (P3) were detached with 0.05 % trypsin [4, 5], rinsed with PBS, and resuspended in stromal medium following cell quantification. Cells suspended in stromal medium were loaded onto scaffolds with a perfusion bioreactor system [27] (Fig. 1). Each system was composed of two bioreactors connected to each other on their lowermost ends and to a peristaltic pump (Ismatec 404b, Glattbrugg, Switzerland) on their uppermost ends with 4.8 mm inner diameter flexible tubing (Tygon, Compagnie de Saint-Gobain, Courbevoie, France). There were three way stopcocks at the top of each bioreactor and 0.22 μm syringe filters (Millipore, Billerica, MA, USA) between tubing attached to the bioreactor and tubing attached to the pump. One bioreactor in each system had a stopcock at its lowermost end.

Photograph of the bioreactor system and a collagen scaffold within an individual bioreactor chamber viewed from the top (upper inset) and side (lower inset). A bioreactor chamber within the system is indicated within a small grey rectangle. The hydrated scaffolds assumed the shape of the bioreactor chambers, and the relative position and orientation was maintained by close approximation of the scaffold perimeter with the chamber walls
Fluid flow direction and rate were controlled by a computer program (LabVIEW, v8.2, National Instruments Corporation, Austin, TX, USA). Scaffolds were hydrated in medium and then placed about 5 mm from the low end of each bioreactor such that fluid flowed only along the short axis. The hydrated scaffolds assumed the shape of the bioreactor chambers, and the relative position and orientation was maintained by close approximation of the scaffold perimeter with the chamber walls. Some minimal vertical displacement was possible during perfusion (~1–3 mm), but orientation was maintained. Systems were primed with stromal medium (5 ml) through the lowermost stopcock followed by addition of MSC suspensions (3 ml, 1 × 106 cells) through the stopcock on the top of each bioreactor. Perfusion was performed for 2 h (37 ºC, 5 % CO2) at a fluid flow rate of 1.05 ml/min (0.97 mm/s). Flow rate was selected based on preliminary tests to optimal cell loading for the system, scaffold and cells [23, 28]. Flow direction was reversed after medium reached the top of a bioreactor so that scaffolds remained immersed.
Following loading, scaffolds were removed, medium collected and the system rinsed with 4 ml of 0.05 % trypsin (Hyclone). Medium and trypsin were centrifuged (350 g, 5 min), cells quantified by hemocytometer and the percent loading efficiency calculated as 100(1−[(cells in medium + cells in trypsin)/initial cell number]). After loading, scaffolds were evaluated or maintained in culture medium according to the study design.
Viable Cell Distribution–Confocal Laser Microscopy
Constructs were incubated in darkness for 30 min at room temperature with a live/dead stain (LIVE/DEAD, Molecular Probes, Eugene, OR, USA) such that live cells fluoresced green while non-viable cells fluoresced red. A total of 20 consecutive photomicrographs (10×) of the entire scaffold cross-section along the short axis were generated every 100 μm (uppermost to lowermost surface) using a spectral confocal laser scanning microscope digital imaging system (Leica TCS SP2, Leica Microsystems, Buffalo Grove, IL, USA). Images were generated from the uppermost surface of the scaffold relative to the position in the bioreactor to the lowermost surface. Graphics software (ImagePro® Media Cybernetics, Bethesda, MF, USA) boundary conditions were set according to the average pixel number (red or green), corresponding to non-viable and viable cells, respectively. Numbers of viable and non-viable cells in each image were estimated based on the number of green and red pixels. The percentage of viable cells was calculated from n = 20 images taken at the center (n = 10) and at the periphery (n = 10). The spatial uniformity ratio was then calculated as % viable surface cells/% viable center cells. This measure was included in the study design to quantify viable cell distribution [29]. The spatial uniformity was evaluated by comparison to completely homogeneous distribution of viable cells across the peripheral and central regions (ratio of 1). Values >1 indicate a larger percentage of viable cells near the scaffold surface, and values of <1 indicate a larger percentage of viable cells near the center of the scaffold.
Lineage Differentiation–Static Culture Conditions
Constructs in 24 well plates (CellStar, BioExpress, Kaysville, UT, USA) were maintained for 7, 14 or 21 days in stromal or differentiation medium with media changes every 3 days (Table 1). For adipogenesis, constructs were initially cultured for 6 days in induction medium followed by maintenance medium.
Microstructure–Light Microscopy
Scaffolds were fixed in 4 % neutral buffered formalin, paraffin embedded, sectioned (5 μm) and stained with Masson’s trichrome. Samples cultured in osteogenic or chondrogenic medium were additionally stained with von Kossa stain (calcium) and alcian blue (proteoglycan), respectively.
Cell Number–DNA Quantity
Constructs stored in TRIReagent (Molecular Research Center, Inc., Cincinatti, OH, USA) at −80 °C after culture were thawed at room temperature (1 min), homogenized (TissueLyser II, Qiagen, Valencia, CA, USA), and DNA and total RNA were isolated by phenol-chloroform extraction [30]. Spectrophotometric (NanoDrop ND-1000, Thermo Scientific, Wilmington, DE, USA) quantification of DNA was performed. The DNA concentration was in addition determined from the 260/280 nm optical density ratio [31, 32]. The total cell number in each scaffold was determined on standard curves of DNA quantity versus numbers of ASC and BMSC [33].
Target Gene Expression–qRT-PCR (Quantitative Reverse Transcription-Polymerase Chain Reaction) mRNA Levels
Following DNAase (Sigma) treatment, RNA yields were determined spectrophotometrically (Nano-drop® ND-1000, Wilmington, DE, USA). RNA was reverse-transcribed to complimentary DNA using oligo(dT) primers (Sigma) and Moloney murine leukemia virus reverse transcriptase (Sigma). Equine primers were designed for MSC (homeobox protein Nanog [Nanog], sex determining region Y-box 2 [Sox2]), as well as adipogenic (peroxisome proliferator-activated receptor gamma [PPARγ], leptin), osteogenic (COLI, alkaline phosphatase [ALP]) and chondrogenic (collagen type II [COLII], sex determining region Y- box 9 [SOX9]) differentiation using Primer3Plus (http://www.primer3plus.com/cgi-bin/dev/primer3plus.cgi) (Table 2).
Target gene mRNA levels were quantified with qRT-PCR using SYBR Green technology (Bio-Rad, Hercules, CA, USA) and an ABI Prism 7900HT sequence detection system (Applied Biosystems, Carlsbad, CA, USA). The threshold cycle (Ct) value of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was subtracted from that of each target gene to determine ΔCt. Lower ΔCt values are consistent with higher mRNA levels while lower mRNA levels have higher ΔCt values. In the case of target gene mRNA signals that cross the threshold earlier than reference gene mRNA levels, the ΔCt value is negative. The more negative the ΔCt value, the higher the target gene mRNA levels.
Ultrastructure–SEM
After culture, constructs were fixed in 2 % paraformaldehyde and 1.25 % glutaraldehyde in 0.1 M sodium cacodylate buffer [26]. Samples were dehydrated in a series of ethanol-distilled water solutions, critical point dried (Polaron, Quorum Technologies, ltd, New Haven, East Sussex, UK) and sputter coated with gold and palladium. They were imaged with SEM (Quanta 200, FEI Company, Hillsboro, OR, USA) at 15 kVP.
Statistical Analysis
Outcomes are reported as mean ± standard error of the mean (SEM). Two replicates for each of the four horse donors were available for each outcome. All response variables were considered continuous and tested for normality using the Shapiro-Wilk statistic. Differences in loading efficiency between cell types were assessed with paired Student’s t-tests within horse. Three- and two-way ANOVA models were used to determine fixed effects of cell type, culture time and induction medium on cell viability and fixed effects of cell type and culture time on mRNA expression levels, respectively. The variation between individual horses was included as the error term for analysis of fixed effects. Significant differences were determined with Tukey’s post-hoc tests (P < 0.05).
Results
Immunophenotype–Flow Cytometry
In combined aliquots of P3 ASC and BMSC harvested from all horses, 99 % of ASC and 90 % of BMSC expressed CD29, 99 % of ASC and 99 % of BMSC expressed CD 44 and 87 % of ASC and 64 % of BMSC expressed CD105 (Fig. 2).

Representative immunophenotype of equine adult equine adipose–(ASC, upper) and bone marrow (BMSC, lower) derived multipotent stromal cells loaded onto collagen type I scaffolds. Black curves represent stained cells and green curves autofluorescence
Loading Efficiency
Loading efficiencies were not significantly different between BMSC (74.6 ± 14.4 %) and ASC (85.8 ± 9.20 %).
Viable Cell Distribution–Confocal Laser Microscopy
Spatial uniformity ratio of viable cells was not affected by cell source, medium type or culture period (Fig. 3, upper). In most cases, the mean spatial uniformity ratio values approximated 1, indicating comparable distribution among surface and center scaffold regions. Immediately after loading, viable cells appeared distinct from each other. Numerous small aggregates of viable cells formed following a period of culture (Fig. 3, lower).

Spatial uniformity ratio (mean ± SEM) of viable cells in adipose–(ASC, upper left) and bone marrow (BMSC, upper right) derived multipotent stromal cell–collagen type I (COLI) constructs after 0, 7, 14 or 21 days of culture in adipogenic, chondrogenic, osteogenic or stromal medium. The spatial uniformity ratio of viable cells was not affected by cell source, medium type or culture period. Confocal laser scanning microscopy photomicrographs representative of both ASC-COLI and BMSC-COLI constructs immediately after cell loading (lower left) and after 21 days of culture in stromal medium (lower right) demonstrate cell distribution in the scaffolds. Immediately after loading, viable cells appeared distinct from each other (lower left). Numerous small aggregates of viable cells formed following a period of culture (lower right). (2.5×, calcein/ethidium bromide)
Microstructure–Light Microscopy
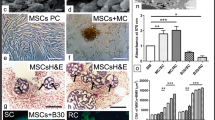
The MSCs on COLI scaffolds cultured in induction medium assumed cell phenotypes consistent with the lineage specificity of the culture medium, and cells were enmeshed in ECM as early as after 7 days of culture. Differences over time were primarily related to cell organization within increasing amounts of ECM. Cells on constructs cultured in adipogenic medium were vacuolated and had an ellipsoid shape (Fig. 4a, b). Intracellular vacuoles were assumed to be the result of adipogenic lipid deposition [34]. Cells in small, loose clusters were present within amorphous ECM. In chondrogenic medium, elongated cells with indistinct nuclei, some within early lacunae, were tightly clustered in a collagenous matrix (Fig. 4c, d) [35–37]. Constructs in osteogenic medium had loose cell clusters of round cells with small, dark nuclei, some within developing lacunae, and spindle shaped cells within collagenous matrix (Fig. 4e, f). Both ASC and BMSC constructs cultured in stromal medium had minimal ECM and cell morphology was consistent with the tissue harvest source (Fig. 4g, h). Based on subjective assessment, the amount of ECM that stained with alcian blue increased in all scaffolds with time of culture in chondrogenic medium (Fig. 5). Von Kossa staining of calcium deposits was evident as early as after 7 days of culture in osteogenic medium (Fig. 5).

Photomicrographs of adult equine bone marrow- (BMSC; a, c, e, g) and adipose (ASC; b, d, f, h) derived multipotent stromal cell–collagen type I (COLI) constructs after 21 days of culture in adipogenic (a, b), chondrogenic (c, d), osteogenic (e, f) or stromal medium (g, h). Cells (a, b; black arrows) on constructs cultured in adipogenic medium were vacuolated and had an ellipsoid shape. Cells (a, b; black arrows) in small, loose clusters were present within amorphous ECM (a, b; red arrows). In chondrogenic medium, elongated cells with indistinct nuclei (c, d; black arrows), some within early lacunae (c, d; asterisks), were tightly clustered in a collagenous matrix (c, d; red arrows). Constructs in osteogenic medium (e, f) had loose cell clusters of round cells with small, dark nuclei (f, black arrow), some within developing lacunae (e; asterisk), and spindle shaped cells (e, black arrow) within collagenous matrix (e, f; red arrows). Both ASC-COLI and BMSC-COLI constructs cultured in stromal medium had minimal extra-cellular matrix (g, h; red arrows) and cell morphology (g, h; black arrows) was consistent with the tissue harvest source, bone marrow (g) or adipose (h). Scaffold remnants were evident in all images (a–h; yellow arrows). Cells (black arrows), new extracellular matrix (red arrows), scaffold remnants (yellow arrows) and lacunae in various stages of development (asterisks) are shown. (100×, Masson’s trichrome stain, scale bar = 10 μm)

Light photomicrographs of adult equine adipose (ASC) and bone marrow- (BMSC) derived multipotent stromal cell–collagen type I constructs cultured for 7, 14 or 21 days in chondrogenic (left two columns, alcian blue stain, 63×) or osteogenic (right two columns, von Kossa stain, 63×) medium. In constructs cultured in chondrogenic medium, cells (left two columns; black arrows) and new extra-cellular matrix (left two columns; red arrows) are evident. In those cultured in osteogenic medium, cells (right two columns; black arrows) and von Kossa staining of calcium deposits (right two columns; blue arrows) are visible. Based on subjective assessment, the amount of extra-cellular matrix that stained with alcian blue increased in all scaffolds with time of culture in chondrogenic medium. Von Kossa staining of calcium deposits was evident as early as after 7 days of culture in osteogenic medium
Cell Number–DNA Quantity
Culture time and medium type affected ASC numbers while only medium type affected BMSC numbers on constructs, and there were no differences attributable to cell source. There were significantly more cells in BMSC constructs cultured in chondrogenic medium than in adipogenic (P < 0.05) or stromal (P < 0.01) medium after 21 days of culture (Fig. 6). The ASC constructs in stromal medium had significantly higher cell numbers than those in induction medium after 21 days (P < 0.001), and ASC constructs cultured in stromal medium had significantly more cells after 21 versus 7 (P < 0.001) and 14 (P < 0.01) days of culture.

Number (mean ± standard error of the mean) of cells adult equine adipose- (ASC; left) and bone marrow- (BMSC; right) derived multipotent stromal cell–collagen type I constructs after 7, 14 or 21 days of culture in adipogenic, chondrogenic, osteogenic or stromal medium. * on columns = Significant differences from day 7 within culture medium cohorts. * above columns = Significant differences between culture media within time points (P < 0.05)
Target Gene Expression–qRT-PCR mRNA Levels
All of the following results were statistically significant. Gene expression after 14 and 21 days of culture are compared to 7 days of culture. Levels of PPARγ were higher in ASC- versus BMSC constructs after 14 days of culture in adipogenic medium (P < 0.001) and LEP was higher after 7 (P < 0.001) and 14 (P < 0.001) days of culture (Fig. 7a, b). The PPARγ mRNA levels decreased in both constructs after 14 (ASC P < 0.01; BMSC P < 0.001) and 21 (ASC P < 0.01; BMSC P < 0.01) days of culture and LEP mRNA increased in BMSC constructs after 14 days (P < 0.001) and in both constructs after 21 days of culture (ASC P < 0.001; BMSC P < 0.001). In chondrogenic medium, construct COLII and SOX9 mRNA levels were higher in BMSC versus ASC constructs at all time points (P < 0.001) with the exception of COLII after 21 days (Fig. 7c, d). Levels of COLII increased in BMSC- and ASC constructs after 14 (ASC P < 0.001; BSMC P < 0.001) and 21 (ASC P < 0.001; BSMC P < 0.001) days of culture. Similarly, levels of SOX 9 also increased in BMSC- and ASC constructs after 14 (ASC P < 0.001; BSMC P < 0.001) and 21 (ASC P < 0.01; BSMC P < 0.001) days of culture. For constructs cultured in osteogenic medium, levels of COLI mRNA were higher in BMSC- compared to ASC constructs at all time points (7 days P < 0.05; 14 days P < 0.001; 21 days P < 0.001), and levels increased in BMSC constructs after 14 (P < 0.01) and 21 (P < 0.001) days of culture (Fig. 7e). BMSC construct ALP mRNA levels were higher than ASC after 7 days of culture (p < 0.001) (Fig. 7f). Levels were higher and lower in ASC constructs after 14 (P < 0.01) and 21 (P < 0.001) days of culture, respectively. Levels decreased in BMSC constructs after 14 (P < 0.001) and 21 (P < 0.001) days of culture. There were no significant differences in Nanog or SOX2 mRNA levels between cell types within time points or within cell types between time points in constructs cultured in stromal medium (Fig. 7g, h).

Delta CT (ΔCT) (mean +/− standard error of the mean) for target gene mRNA levels in adult equine bone marrow- (BMSC) and adipose (ASC) derived multipotent stromal cell–collagen type I (COLI) constructs after 7, 14, or 21 days of culture in adipogenic (a, b), chondrogenic (c, d), osteogenic (e, f) or stromal (g, h) medium. * on columns = significant differences between cell types within time points. # above columns = significant differences from day 7 within cell type (P < 0.05). Lower ΔCt values are consistent with higher mRNA levels while lower mRNA levels have higher ΔCt values. In the case of target gene mRNA signals that cross the threshold earlier than reference gene mRNA levels, the ΔCt value is negative. The more negative the ΔCt value, the higher the target gene mRNA levels
Ultrastructure–SEM
Ultrastructure showed lineage-specific cell morphology and ECM (Fig. 8). Constructs in adipogenic medium had round to ellipsoid cells in amorphous matrix. Scaffold constructs in chondrogenic medium had spherical cells embedded in dense, highly organized ECM. Those in osteogenic medium had flattened, polygonal and spindle-shaped cells adhered to collagen fibers and surrounded by mineral deposits [38]. Constructs in stromal medium were characterized by abundant, poorly organized collagen fibrils.

Scanning electron photomicrographs of adult equine bone marrow- (BMSC; a–c) and adipose (ASC; d–f) derived multipotent stromal cell–collagen type I (COLI) constructs cultured in adipogenic (a, d), chondrogenic (b, e) or osteogenic (c, f) medium for 21 days. A scaffold without cells (Scaffold) and with ASC in stromal medium (Stromal) are shown for comparison. Constructs in adipogenic medium had round (a; black arrow) to ellipsoid (d, black arrow) cells in amorphous matrix (a; M) containing lipid droplets (d; white arrows) and collagen fibrils (d; gray arrow head). Scaffold collagen fibers were also visible (a; black arrow head). Scaffold constructs in chondrogenic medium had dense, highly organized extracellular matrix (b, e; M) with abundant collagen fibrils (b, e; gray arrow heads). Those in osteogenic medium had flattened, polygonal cells (c; black arrow) adhered to collagen fibers (c, f; black arrow heads) with mineral deposits (c, f; white arrow heads) and collagen fibrils (c, f; gray arrow heads) in the surrounding matrix. Constructs in stromal medium were characterized by abundant, poorly organized collagen fibrils (Stromal; gray arrow head) surrounding collagen fibers (Stromal; black arrow head). (Scale bars = 10 μm)
Discussion
Comparable mesenchymal trilineage differentiation and ECM production was successfully achieved with equine ASC and BMSC seeded in COLI scaffolds with lineage specific culture media in this study. Cells had similar loading efficiency, viability, and spatial distribution following identical perfusion bioreactor loading and static culture, supporting the first hypothesis. Though ASC tended to exhibit earlier and greater upregulation of adipogenic target genes, and the same was true for BMSC and osteogenic target genes, some differences in mRNA levels between cell types declined with culture time and micro- and ultrastructure was not greatly different between them. Similarly, though BMSC had earlier and higher upregulation of chondrogenic target genes, proteoglycan staining and micro- and ultrastructures were comparable between ASC and BMSC. Contradictory to the second hypothesis, these results confirm that the capability of cryopreserved MSC for adipogenic, osteogenic and chondrogenic differentiation and ECM production did not significantly depend on tissue source.
Perfusion bioreactor seeding of COLI scaffolds used in this study was a reliable, repeatable mechanism to load and distribute adult equine ASC and BMSC into COLI scaffolds at a cell density (~5 × 106 cells/cm3) within the range of those reported for mesenchymal tissue generation [14, 15, 39]. Cell seeding by repeated perfusion through scaffold enhances cell number and distribution, which in turn promote uniform tissue generation and reduce time required for the generation process itself [21, 23]. Optimal fluid flow rate is uniquely determined by scaffold microstructure; velocities that are too high interfere with cell adhesion and those that are too low do not move cells through the system or distribute them throughout the scaffold [23, 27, 40]. As described in “Materials and Methods”, the flow rate was selected based on preliminary work. While further optimization may be possible, the perfusion rate used in this study resulted in loading efficiencies that are consistent with comparable studies [21, 23, 27].
The numbers of cells on the scaffolds based on DNA content remained relatively stable with the exception of a large increase in ASC constructs cultured in stromal medium and gradual increase in BMSC constructs in chondrogenic medium. These findings may have been due to continued undifferentiated cell proliferation [41] and cell proliferation accompanying chondrogenic differentiation [42, 43], respectively. The otherwise stable cell numbers support the observed cell differentiation [44].
Culture of three-dimensional MSC-scaffold constructs in perfusion bioreactors likely provides more favorable and physiologically comparable conditions than the static culture conditions used in this study [23, 27, 40]. The continuous flow through perfusion bioreactor systems allows dynamic nutrient supply, gas exchange and waste removal. Additionally, fluid perfusion helps to maintain similar local environments in surface and center scaffold regions. This point is especially relevant for larger engineered tissue constructs that have smaller surface area to volume ratios. It was not possible to maintain the large number of constructs required for this study in available bioreactors for the extended culture periods within the study period. Despite this potential limitation, the constructs had ECM formation and viable cells throughout, indicating that static culture with regular media changes was sufficient to support cell viability and differentiation. This finding has been previously reported for MSC on comparably sized COLI scaffolds [14]. However, dynamic perfusion may provide a physiochemical environment that is more inductive to tissue formation with optimum nutrient delivery, gas exchange and waste removal in larger constructs [40].
The ability of equine ASC and BMSC to differentiate into adipogenic, chondrogenic and osteogenic lineages is established [4, 5, 8]. Though there is no clear agreement, MSC appear to have a propensity to assume their lineage of origin under standard induction conditions, potentially due to epigenetic programming [45–48]. Tissue specific target gene upregulation by ASC- and BMSC-COLI constructs in this investigation generally support this concept, though the micro- and ultrastructure did not appear different between ASC and BMSC constructs. The mRNA levels tended to increase with induction in all scaffolds with the exception of PPARγ and ALP, which decreased as expected with cell maturation [35, 48, 49]. Epigenetic processes may support a more rapid response of adult MSCs to induction media of their tissue of origin [50]. However, it is conceivable that effects are less profound with respect to ECM production since genetic upregulation based on mRNA levels does not necessarily directly correlate with protein formation central to tissue engineering [42, 51]. These results support comparable mesenchymal trilineage differentiation and ECM forming capabilities of equine BMSC and ASC on COLI scaffolds. Longer in vitro culture periods and controlled in vivo comparisons may help reveal differences in quality and function of mature tissues from different cell sources.
The donor population in this study was selected to eliminate variability from age, gender or breed. The MSC phenotype was confirmed by expression of cell surface antigens [52–54]. Relatively low CD105 expression relative to CD29 and CD44 by MSCs has been previously reported in several species, including equine [54]. Taken together, the results support an MSC phenotype from both tissue sources used in this investigation.
Scaffolds composed of COLI were used in this investigation for comparison between MSC to produce tissue specific matrix directed only by induction media on a ubiquitous component of mesenchymal ECM [12, 13]. Scaffolds are often customized for specific tissue generation by addition of tissue specific growth factors or ECM proteins, among other components [55]. As indicated above, we sought to establish the potential of ASC and BMSC to differentiate and produce mesenchymal ECM without scaffold alterations. It is possible that scaffolds tailored to cell type and tissue target may enhance the mesenchymal ECM forming potential seen in this investigation.
Xenogeneic collagen implants have the potential to be antigenic and immunogenic. There is a report of human humoral responses to a commercially available bovine COLI implant, ZYDERM® [56]. The physician package insert for the product indicates that fewer than 1 % of recipients have self-limiting, local inflammatory reactions that may persist throughout the life of the implant. According to the insert, the reactions do not affect the long-term success of the collagen implant corrections. An in vitro study showed that porcine and human COLI elicit proliferation of human immune cell subpopulations in co-stimulation with anti-CD3 antibody with accompanying release of cytokines interferon gamma and tumor necrosis factor alpha [57]. A more recent in vitro study showed that xenogeneic COLI did not trigger maturation of human dendritic cells and had no effect on B or T cell proliferation or cytokine response [58]. Further, a previous study showed that the local inflammatory reaction to scaffolds composed of β-tricalcium phosphate and bovine COLI was reduced by implantation with syngeneic or allogeneic ASCs in a rat spinal fusion model [59]. Finally, a recent report indicates that ovine amnionic epithelial cell xenografts in naturally occurring equine superficial digital flexor tendon injuries produced ovine COLI without apparent rejection [60]. Given potential immunogenicity of COLI xenografts, preclinical and clinical outcome assessments should include evaluations of local and systemic responses with the understanding that there is potential for immune stimulation.
There are a number of approaches to MSC-scaffold tissue formation, two of which include implantation of constructs after a short in vitro incubation for cell attachment or orthotopic placement after one to several weeks of in vitro culture in induction medium for MSC differentiation and ECM formation [61]. A potential limitation of the former is reliance on the recipient microenvironment to direct cell differentiation and ECM formation [62]. Microenvironmental changes from local or systemic pathology may disrupt normal MSC function and differentiation [63]. In contrast, constructs that contain differentiated cells and tissue-specific ECM from pre-implantation culture in induction medium may serve as a source of growth factors to recruit and direct native progenitor cells as well as to provide an ECM matrix for tissue conduction and engraftment [64]. It is imperative to fully characterize in vitro MSC behavior in scaffolds to identify the best combinations and preimplantation culture conditions prior to in vivo implantation.
The comparable abilities of cryopreserved adult equine ASC and BMSC for adipogenic, osteogenic and chondrogenic differentiation and ECM production on COLI scaffolds in vitro following perfusion bioreactor loading and culture in this study is vital to efforts toward translation of MSC tissue engineering to equine patients. In vivo validation of the findings through preclinical models prior to controlled clinical trials will continue the momentum toward a goal of customized, viable tissue implants to augment fracture, wound and intra-articular therapies.
References
Carstanjen, B., Desbois, C., Hekmati, M., & Behr, L. (2006). Successful engraftment of cultured autologous mesenchymal stem cells in a surgically repaired soft palate defect in an adult horse. Canadian Journal of Veterinary Research, 70, 143–147.
Pacini, S., Spinabella, S., Trombi, L., Fazzi, R., Galimberti, S., Dini, F., et al. (2007). Suspension of bone marrow-derived undifferentiated mesenchymal stromal cells for repair of superficial digital flexor tendon in race horses. Tissue Engineering, 13, 2949–2955.
Colleoni, S., Bottani, E., Tessaro, I., Mari, G., Merlo, B., Romagnoli, N., et al. (2009). Isolation, growth and differentiation of equine mesenchymal stem cells: effect of donor, source, amount of tissue and supplementation with basic fibroblast growth factor. Veterinary Research Communications, 33, 811–821.
Vidal, M. A., Kilroy, G. E., Johnson, J. R., Lopez, M. J., Moore, R. M., & Gimble, J. M. (2006). Cell growth characteristics and differentiation frequency of adherent equine bone marrow-derived mesenchymal stromal cells: adipogenic and osteogenic capacity. Veterinary Surgery, 35, 601–610.
Vidal, M. A., Kilroy, G. E., Lopez, M. J., Johnson, J. R., Moore, R. M., & Gimble, J. M. (2007). Characterization of equine adipose tissue-derived stromal cells: adipogenic and osteogenic capacity and comparison with bone marrow-derived mesenchymal stromal cells. Veterinary Surgery, 36, 613–622.
Schnabel, L. V., Lynch, M. E., van der Meulen, M. C., Yeager, A. E., Kornatowski, M. A., & Nixon, A. J. (2009). Mesenchymal stem cells and insulin-like growth factor-I gene-enhanced mesenchymal stem cells improve structural aspects of healing in equine flexor digitorum superficialis tendons. Journal of Orthopaedic Research, 27, 1392–1398.
Mensing, N., Gasse, H., Hambruch, N., Haeger, J. D., Pfarrer, C., & Staszyk, C. (2011). Isolation and characterization of multipotent mesenchymal stromal cells from the gingiva and the periodontal ligament of the horse. BMC Veterinary Research, 7, 42.
Kon, E., Mutini, A., Arcangeli, E., Delcogliano, M., Filardo, G., Nicoli Aldini, N., et al. (2010). Novel nanostructured scaffold for osteochondral regeneration: pilot study in horses. Journal of Tissue Engineering and Regenerative Medicine, 4, 300–308.
Guaccio, A., Guarino, V., Perez, M. A., Cirillo, V., Netti, P. A., & Ambrosio, L. (2011). Influence of electrospun fiber mesh size on hMSC oxygen metabolism in 3D collagen matrices: experimental and theoretical evidences. Biotechnology and Bioengineering, 108, 1965–1976.
Karageorgiou, V., & Kaplan, D. (2005). Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials, 26, 5474–5491.
Karande, T. S., Ong, J. L., & Agrawal, C. M. (2004). Diffusion in musculoskeletal tissue engineering scaffolds: design issues related to porosity, permeability, architecture, and nutrient mixing. Annals of Biomedical Engineering, 32, 1728–1743.
Chang, S. C., Chung, H. Y., Tai, C. L., Chen, P. K., Lin, T. M., & Jeng, L. B. (2010). Repair of large cranial defects by hBMP-2 expressing bone marrow stromal cells: comparison between alginate and collagen type I systems. Journal of Biomedical Materials Research. Part A, 94, 433–441.
Davidenko, N., Campbell, J. J., Thian, E. S., Watson, C. J., & Cameron, R. E. (2010). Collagen-hyaluronic acid scaffolds for adipose tissue engineering. Acta Biomaterialia, 6, 3957–3968.
Akkouch, A., Zhang, Z., & Rouabhia, M. (2011). A novel collagen/hydroxyapatite/poly(lactide-co-epsilon-caprolactone) biodegradable and bioactive 3D porous scaffold for bone regeneration. Journal of Biomedical Materials Research. Part A, 96, 693–704.
Chen, W. C., Yao, C. L., Wei, Y. H., & Chu, I. M. (2011). Evaluating osteochondral defect repair potential of autologous rabbit bone marrow cells on type II collagen scaffold. Cytotechnology, 63, 13–23.
Welsch, G. H., Mamisch, T. C., Zak, L., Blanke, M., Olk, A., Marlovits, S., et al. (2010). Evaluation of cartilage repair tissue after matrix-associated autologous chondrocyte transplantation using a hyaluronic-based or a collagen-based scaffold with morphological MOCART scoring and biochemical T2 mapping: preliminary results. The American Journal of Sports Medicine, 38, 934–942.
Engelhardt, E. M., Micol, L. A., Houis, S., Wurm, F. M., Hilborn, J., Hubbell, J. A., et al. (2011). A collagen-poly(lactic acid-co-varepsilon-caprolactone) hybrid scaffold for bladder tissue regeneration. Biomaterials, 32, 3969–3976.
Shi, C., Li, Q., Zhao, Y., Chen, W., Chen, B., Xiao, Z., et al. (2011). Stem-cell-capturing collagen scaffold promotes cardiac tissue regeneration. Biomaterials, 32, 2508–2515.
Sarkar, A., Tatlidede, S., Scherer, S. S., Orgill, D. P., & Berthiaume, F. (2011). Combination of stromal cell-derived factor-1 and collagen-glycosaminoglycan scaffold delays contraction and accelerates reepithelialization of dermal wounds in wild-type mice. Wound Repair and Regeneration, 19, 71–79.
Bernemann, I., Mueller, T., Blasczyk, R., Glasmacher, B., & Hofmann, N. (2011). Colonization of collagen scaffolds by adipocytes derived from mesenchymal stem cells of the common marmoset monkey. Biochemical and Biophysical Research Communications, 411, 317–322.
Koch, M. A., Vrij, E. J., Engel, E., Planell, J. A., & Lacroix, D. (2010). Perfusion cell seeding on large porous PLA/calcium phosphate composite scaffolds in a perfusion bioreactor system under varying perfusion parameters. Journal of Biomedical Materials Research. Part A, 95, 1011–1018.
Wendt, D., Marsano, A., Jakob, M., Heberer, M., & Martin, I. (2003). Oscillating perfusion of cell suspensions through three-dimensional scaffolds enhances cell seeding efficiency and uniformity. Biotechnology and Bioengineering, 84, 205–214.
Zhao, F., & Ma, T. (2005). Perfusion bioreactor system for human mesenchymal stem cell tissue engineering: dynamic cell seeding and construct development. Biotechnology and Bioengineering, 91, 482–493.
Radisic, M., Euloth, M., Yang, L., Langer, R., Freed, L. E., & Vunjak-Novakovic, G. (2003). High-density seeding of myocyte cells for cardiac tissue engineering. Biotechnology and Bioengineering, 82, 403–414.
Martin, I., Wendt, D., & Heberer, M. (2004). The role of bioreactors in tissue engineering. Trends in Biotechnology, 22, 80–86.
Vasanjee, S. C., Paulsen, D., Hosgood, G., Robinson, S. O., & Lopez, M. J. (2008). Characterization of normal canine anterior cruciate ligament-associated synoviocytes. Journal of Orthopaedic Research, 26, 809–815.
Maidhof, R., Marsano, A., Lee, E. J., & Vunjak-Novakovic, G. (2010). Perfusion seeding of channeled elastomeric scaffolds with myocytes and endothelial cells for cardiac tissue engineering. Biotechnology Progress, 26, 565–572.
Gaspar, D. A., Gomide, V., & Monteiro, F. J. (2012). The role of perfusion bioreactors in bone tissue engineering. Biomatter, 2, 167–175.
Kennedy, J. P., McCandless, S. P., Rauf, A., Williams, L. M., Hillam, J., & Hitchcock, R. W. (2011). Engineered channels enhance cellular density in perfused scaffolds. Acta Biomaterialia, 7, 3896–3904.
Rio, D. C., Ares, M., Jr., Hannon, G. J., Nilsen, T. W. (2010). Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protocol, 2010, pdb prot5439.
Lee, J. H., Park, Y., Choi, J. R., Lee, E. K., & Kim, H. S. (2010). Comparisons of three automated systems for genomic DNA extraction in a clinical diagnostic laboratory. Yonsei Medical Journal, 51, 104–110.
Radpour, R., Sikora, M., Grussenmeyer, T., Kohler, C., Barekati, Z., Holzgreve, W., et al. (2009). Simultaneous isolation of DNA, RNA, and proteins for genetic, epigenetic, transcriptomic, and proteomic analysis. Journal of Proteome Research, 8, 5264–5274.
Haque, K. A., Pfeiffer, R. M., Beerman, M. B., Struewing, J. P., Chanock, S. J., & Bergen, A. W. (2003). Performance of high-throughput DNA quantification methods. BMC Biotechnology, 3, 20.
Venugopal, B., Fernandez, F. B., Babu, S. S., Harikrishnan, V. S., Varma, H., & John, A. (2012). Adipogenesis on biphasic calcium phosphate using rat adipose-derived mesenchymal stem cells: in vitro and in vivo. Journal of Biomedical Materials Research. Part A, 100, 1427–1437.
Liu, F., Malaval, L., & Aubin, J. E. (2003). Global amplification polymerase chain reaction reveals novel transitional stages during osteoprogenitor differentiation. Journal of Cell Science, 116, 1787–1796.
Nurminsky, D., Shanmugasundaram, S., Deasey, S., Michaud, C., Allen, S., Hendig, D., et al. (2011). Transglutaminase 2 regulates early chondrogenesis and glycosaminoglycan synthesis. Mechanisms of Development, 128, 234–245.
Steinert, A. F., Proffen, B., Kunz, M., Hendrich, C., Ghivizzani, S. C., Noth, U., et al. (2009). Hypertrophy is induced during the in vitro chondrogenic differentiation of human mesenchymal stem cells by bone morphogenetic protein-2 and bone morphogenetic protein-4 gene transfer. Arthritis Research & Therapy, 11, R148.
Qian, X., Yuan, F., Zhimin, Z., Anchun, M. (2013). Dynamic perfusion bioreactor system for 3D culture of rat bone marrow mesenchymal stem cells on nanohydroxyapatite/polyamide 66 scaffold in vitro. Journal of Biomedical Materials Research Part B: Applied Biomaterials.
Luo, F., Hou, T. Y., Zhang, Z. H., Xie, Z., Wu, X. H., & Xu, J. Z. (2013). Effects of initial cell density and hydrodynamic culture on osteogenic activity of tissue-engineered bone grafts. PLoS One, 8, e53697.
Janssen, F. W., van Dijkhuizen-Radersma, R., Van Oorschot, A., Oostra, J., de Bruijn, J. D., & Van Blitterswijk, C. A. (2010). Human tissue-engineered bone produced in clinically relevant amounts using a semi-automated perfusion bioreactor system: a preliminary study. Journal of Tissue Engineering and Regenerative Medicine, 4, 12–24.
Gustafsson, Y., Haag, J., Jungebluth, P., Lundin, V., Lim, M. L., Baiguera, S., et al. (2012). Viability and proliferation of rat MSCs on adhesion protein-modified PET and PU scaffolds. Biomaterials, 33, 8094–8103.
Murdoch, A. D., Grady, L. M., Ablett, M. P., Katopodi, T., Meadows, R. S., & Hardingham, T. E. (2007). Chondrogenic differentiation of human bone marrow stem cells in transwell cultures: generation of scaffold-free cartilage. Stem Cells, 25, 2786–2796.
Dexheimer, V., Frank, S., & Richter, W. (2012). Proliferation as a requirement for in vitro chondrogenesis of human mesenchymal stem cells. Stem Cells and Development, 21, 2160–2169.
Park, S. B., Seo, K. W., So, A. Y., Seo, M. S., Yu, K. R., Kang, S. K., et al. (2012). SOX2 has a crucial role in the lineage determination and proliferation of mesenchymal stem cells through Dickkopf-1 and c-MYC. Cell Death and Differentiation, 19, 534–545.
Eilertsen, K. J., Floyd, Z., & Gimble, J. M. (2008). The epigenetics of adult (somatic) stem cells. Critical Reviews in Eukaryotic Gene Expression, 18, 189–206.
Sakaguchi, Y., Sekiya, I., Yagishita, K., & Muneta, T. (2005). Comparison of human stem cells derived from various mesenchymal tissues: superiority of synovium as a cell source. Arthritis and Rheumatism, 52, 2521–2529.
Pachon-Pena, G., Yu, G., Tucker, A., Wu, X., Vendrell, J., Bunnell, B. A., et al. (2011). Stromal stem cells from adipose tissue and bone marrow of age-matched female donors display distinct immunophenotypic profiles. Journal of Cellular Physiology, 226, 843–851.
Wilke, M. M., Nydam, D. V., & Nixon, A. J. (2007). Enhanced early chondrogenesis in articular defects following arthroscopic mesenchymal stem cell implantation in an equine model. Journal of Orthopaedic Research, 25, 913–925.
Fink, T., Abildtrup, L., Fogd, K., Abdallah, B. M., Kassem, M., Ebbesen, P., et al. (2004). Induction of adipocyte-like phenotype in human mesenchymal stem cells by hypoxia. Stem Cells, 22, 1346–1355.
Berdasco, M., & Esteller, M. (2011). DNA methylation in stem cell renewal and multipotency. Stem Cell Research & Therapy, 2, 42.
Lossdorfer, S., Gotz, W., & Jager, A. (2005). PTH(1-34) affects osteoprotegerin production in human PDL cells in vitro. Journal of Dental Research, 84, 634–638.
Lopez, M. J., & Daigle, P. R. (2013). Adult multipotent stromal cell technology for bone regeneration: a review. Veterinary Surgery, 42, 1–11.
Radcliffe, C. H., Flaminio, M. J., & Fortier, L. A. (2010). Temporal analysis of equine bone marrow aspirate during establishment of putative mesenchymal progenitor cell populations. Stem Cells and Development, 19, 269–282.
De Schauwer, C., Piepers, S., Van de Walle, G. R., Demeyere, K., Hoogewijs, M. K., Govaere, J. L., et al. (2012). In search for cross-reactivity to immunophenotype equine mesenchymal stromal cells by multicolor flow cytometry. Cytometry. Part A, 81, 312–323.
Lin, H., Yang, G., Tan, J., & Tuan, R. S. (2012). Influence of decellularized matrix derived from human mesenchymal stem cells on their proliferation, migration and multi-lineage differentiation potential. Biomaterials, 33, 4480–4489.
Ellingsworth, L. R., DeLustro, F., Brennan, J. E., Sawamura, S., & McPherson, J. (1986). The human immune response to reconstituted bovine collagen. Journal of Immunology, 136, 877–882.
Bayrak, A., Tyralla, M., Ladhoff, J., Schleicher, M., Stock, U. A., Volk, H. D., et al. (2010). Human immune responses to porcine xenogeneic matrices and their extracellular matrix constituents in vitro. Biomaterials, 31, 3793–3803.
Bayrak, A., Pruger, P., Stock, U. A., & Seifert, M. (2013). Absence of immune responses with xenogeneic collagen and elastin. Tissue Engineering. Part A, 19, 1592–1600.
Lopez, M. J., McIntosh, K. R., Spencer, N. D., Borneman, J. N., Horswell, R., Anderson, P., et al. (2009). Acceleration of spinal fusion using syngeneic and allogeneic adult adipose derived stem cells in a rat model. Journal of Orthopaedic Research, 27, 366–373.
Muttini, A., Valbonetti, L., Abate, M., Colosimo, A., Curini, V., Mauro, A., et al. (2013). Ovine amniotic epithelial cells: in vitro characterization and transplantation into equine superficial digital flexor tendon spontaneous defects. Research in Veterinary Science, 94, 158–169.
Caplan, A. I. (2007). Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. Journal of Cellular Physiology, 213, 341–347.
Bajada, S., Mazakova, I., Richardson, J. B., & Ashammakhi, N. (2008). Updates on stem cells and their applications in regenerative medicine. Journal of Tissue Engineering and Regenerative Medicine, 2, 169–183.
Huff, N. K., Spencer, N. D., Gimble, J. M., Bagby, G. J., Nelson, S., & Lopez, M. J. (2011). Impaired expansion and multipotentiality of adult stromal cells in a rat chronic alcohol abuse model. Alcohol, 45, 393–402.
Erickson, I. E., Kestle, S. R., Zellars, K. H., Dodge, G. R., Burdick, J. A., & Mauck, R. L. (2012). Improved cartilage repair via in vitro pre-maturation of MSC-seeded hyaluronic acid hydrogels. Biomedical Materials, 7, 024110.
Acknowledgments
Funding for this study was provided in part by the Grayson Jockey Club Research Foundation, Louisiana State University Equine Health Studies Program and the National Institutes of Health (EB002520).
Competing Interests
No competing interests are declared.
Author information
Authors and Affiliations
Corresponding author
Additional information
Previous Publications
Xie L, Marsano A, Zhang Y, Vunjak-Novakovic G, Lopez MJ. In Vitro Tissue Generation by Adult Equine Multipotent Stromal Cells on Collagen Scaffolds. Proceedings American Association of Equine Practitioners Meeting, 2012.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Xie, L., Zhang, N., Marsano, A. et al. In Vitro Mesenchymal Trilineage Differentiation and Extracellular Matrix Production by Adipose and Bone Marrow Derived Adult Equine Multipotent Stromal Cells on a Collagen Scaffold. Stem Cell Rev and Rep 9, 858–872 (2013). https://doi.org/10.1007/s12015-013-9456-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-013-9456-1