
Abstract
The correct discrimination between native and near-native protein conformations is essential for achieving accurate computer-based protein structure prediction. However, this has proven to be a difficult task, since currently available physical energy functions, empirical potentials and statistical scoring functions are still limited in achieving this goal consistently. In this work, we assess and compare the ability of different full atom knowledge-based potentials to discriminate between native protein structures and near-native protein conformations generated by comparative modeling. Using a benchmark of 152 near-native protein models and their corresponding native structures that encompass several different folds, we demonstrate that the incorporation of close non-bonded pairwise atom terms improves the discriminating power of the empirical potentials. Since the direct and unbiased derivation of close non-bonded terms from current experimental data is not possible, we obtained and used those terms from the corresponding pseudo-energy functions of a non-local knowledge-based potential. It is shown that this methodology significantly improves the discrimination between native and near-native protein conformations, suggesting that a proper description of close non-bonded terms is important to achieve a more complete and accurate description of native protein conformations. Some external knowledge-based energy functions that are widely used in model assessment performed poorly, indicating that the benchmark of models and the specific discrimination task tested in this work constitutes a difficult challenge.






Similar content being viewed by others
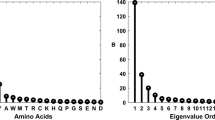
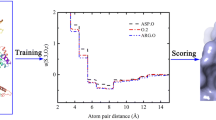

References
Sippl, M. J. (1993). Boltzmann’s principle, knowledge-based mean fields and protein folding. An approach to the computational determination of protein structures. Journal of Computer Aided Molecular Design, 7, 473–501.
Ponder, J. W., & Case, D. A. (2003). Force fields for protein simulations. Advances in Protein Chemistry, 66, 27–85.
Sippl, M. J. (1990). Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. Journal of Molecular Biology, 213, 859–883.
Melo, F., Sanchez, R., & Sali, A. (2002). Statistical potentials for fold assessment. Protein Science, 11, 430–448.
Lazaridis, T., & Karplus, M. (1998). Discrimination of the native from misfolded protein models with an energy function including implicit solvation. Journal of Molecular Biology, 288, 477–487.
Samudrala, R., & Moult, J. (1998). An all-atom distance-dependent conditional probability discriminatory function for protein structure prediction. Journal of Molecular Biology, 275, 895–916.
Zhou, H., & Zhou, Y. (2002). Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Science, 11, 2714–2726.
Lu, H., & Skolnick, J. (2001). A distance-dependent atomic knowledge-based potential for improved protein structure selection. Proteins, 44, 223–232.
Melo, F., & Feytmans, E. (1997). Novel knowledge-based mean force potential at atomic level. Journal of Molecular Biology, 267, 207–222.
Melo, F., & Feytmans, E. (1998). Assessing protein structures with a non-local atomic interaction energy. Journal of Molecular Biology, 277, 1141–1152.
Melo, F., Devos, D., Depiereux, E., & Feytmans, E. (1997). ANOLEA: a www server to assess protein structures. Intelligent Systems for Molecular Biology, 97, 187–190.
Sali, A., & Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology, 234, 779–815.
Fiser, A., Do, R. K., & Sali, A. (2000). Modeling of loops in protein structures. Protein Science, 9, 1753–1773.
MacKerell, A. D. Jr., Bashford, D., Bellott, M., Dunbrack RJaEJ, Field, M., Fischer, S., Gao, J., Guo, H., Ha, S., et al. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B, 102, 3586–3616.
Brooks, B., Bruccoleri, R., Olafsonand, B., States, D., Swaminathan, S., & Karplus, M. (1983). CHARMM: A program for macromolecular energy, minimizations and dynamic calculations. Journal of Computational Chemistry, 4, 187–217.
Sippl, M. J. (1993). Recognition of errors in three-dimensional structures of proteins. Proteins, 17, 355–362.
Murzin, A. G., Brenner, S. E., Hubbard, T., & Chothia, C. (1995). SCOP: A structural classification of proteins database for the investigation of sequences and structures. Journal of Molecular Biology, 247, 536–540.
Andreeva, A., Howorth, D., Brenner, S. E., Hubbard, T. J., Chothia, C., & Murzin, A. G. (2004). SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Research, 32, 226–229.
Orengo, C. A., Michie, A. D., Jones, S., Jones, D. T., Swindells, M. B., & Thornton, J. M. (1997). CATH: A Hierarchic Classification of Protein Domain Structures. Structure, 5, 1093–1108.
Sanchez, R., & Sali, A. (1998). Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proceedings of National Academy of Sciences of the United States of America, 95, 13597–13602.
Berman, H. M., Battistuz, T., Bhat, T. N., Bluhm, W. F., Bourne, P. E., Burkhardt, K., Feng, Z., Gilliland, G. L., Iype, L., & Jain, S., et al (2002). The Protein Data Bank. Acta Cryst D, 58, 899–907.
Altschul, S. (1998). Generalized affine gap costs for protein sequence alignment. Proteins, 32, 88–96.
Fawcett, T. (2004). ROC graphs: Notes and practical considerations for researchers. Kluwer Academic Publishers, 1, 1–38.
Swets, J. A. (1988). Measuring the accuracy of diagnostic systems. Science, 240, 1285–1293.
Swets, J. A., Dawes, R. M., & Monahan, J. (2000). Better decisions through science. Scientific American, 283, 82–87.
Metz, C. E., Herman, B. A., & Roe, C. A. (1998). Statistical comparison of two ROC curve estimates obtained from partially-paired datasets. Medical Decision Making, 18, 110–121.
Metz, C. E. (1986). ROC methodology in radiological imaging. Investigative Radiology, 21, 720–733.
Metz, C. E., Wang, P. L., & Kronman, H. B. (1984). A new approach for testing the significance of differences between ROC curves measured from correlated data. In F. Deconinck (Ed.), Information processing in medical imaging (pp. 432–445). The Hague: Nijhoff.
Samudrala, R., & Levitt, M. (2000). Decoys ‘R’ Us: A database of incorrect conformations to improve protein structure prediction. Protein Science, 9, 1399–1401.
Hendlich, M., Lackner, P., Weitckus, S., Floeckner, H., Froschauer, R., Gottsbacher, K., Casari, G., & Sippl, M. J. (1990). Identification of native protein folds amongst a large number of incorrect models. Journal of Molecular Biology, 216, 167–180.
Simons, K. T., Ruczinski, I., Kooperberg, C., Fox, B. A., Bystroff, C., & Baker, D. (1999). Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins, 34, 82–95.
Casari, G., & Sippl, M. J. (1992). Hydrophobic potential derived from X-ray structures of globular proteins is able to identify native proteins. Journal of Molecular Biology, 224, 725–732.
Gatchell, D. W., Dennis, S., & Vajda, S. (2000). Discrimination of near-native protein structures from misfolded models by empirical free energy functions. Proteins, 41, 518–534.
Vendruscolo, M., Najmanovich, R., & Domany, E. (2000). Can a pairwise contact potential stabilize native protein folds against decoys obtained by threading? Proteins, 38, 134–148.
Sippl, M. J. (1995). Knowledge-based potentials for proteins. Current Opinion in Structural Biology, 5, 229–235.
Sippl, M. J., & Weitckus, S. (1992). Detection of native like models for amino acid sequences of unknown three dimensional structure in a data base of known protein conformations. Proteins, 13, 258–271.
Solis, A. D., & Rackovsky, S. (2006). Improvement of statistical potentials and threading score functions using information maximization. Proteins, 62, 892–908.
Acknowledgements
This work was funded by grant 1051112 from FONDECYT. We are also grateful to M.S. Madhusudhan for his useful comments and careful reading of this manuscript. Finally, we would like to acknowledge Leonardo Sepúlveda and Tomás Pérez Acle for performing the calculations with CHARMM and OPLS force fields reported in this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ferrada, E., Vergara, I.A. & Melo, F. A Knowledge-Based Potential with an Accurate Description of Local Interactions Improves Discrimination between Native and Near-Native Protein Conformations. Cell Biochem Biophys 49, 111–124 (2007). https://doi.org/10.1007/s12013-007-0050-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-007-0050-5