
Abstract
Recently emerged viral infectious diseases (VIDs) include HIV/AIDS, influenzas H5N1 and 2009 H1N1, SARS, and Ebola hemorrhagic fevers. Earlier research determined metabolic oxidative stress in hosts deficient in antioxidant selenium (Se) (<1 μMol Se/L of blood) induces both impaired human host immunocompetence and rapidly mutated benign variants of RNA viruses to virulence. These viral mutations are consistent, rather than stochastic, and long-lived. When Se-deficient virus-infected hosts were supplemented with dietary Se, viral mutation rates diminished and immunocompetence improved. Herein is described the role of micronutrient Se deficiency on the evolution of some contemporary RNA viruses and their subsequent VIDs. Distinguishing cellular and biomolecular evidence for several VIDs suggests that environmental conditions conducive to chronic dietary Se deprivation could be monitored for bioindicators of incipient viral virulence and subsequent pathogenesis.
Similar content being viewed by others


Introduction
Viral infectious diseases (VIDs), primarily influenzas and human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS), take personal, social, and economic tolls—HIV/AIDS ranks among the five global causes of death now and for the foreseeable future [1]. Influenza, in the United States alone, claims an estimated 36,000 people from annual epidemics and associated respiratory complications [2]; during pandemics, when the population has developed little immunity against novel influenza viruses, this figure can increase. Though socioeconomic/ecological factors contribute to the emergence and spread of various infectious diseases [1–3], a host’s nutritional status was found to influence the etiology of specifically VIDs. This review describes the role of chronic micronutrient selenium (Se) deficiency on the evolution of some contemporary RNA viruses and their subsequent VIDs [4].
Significance of host Se status is based on the antioxidant properties of amino acid (aa) selenocysteine, the catalytic center of selenoenzymes. The glutathione peroxidase (GPx) family regulates biologic oxidative homeostasis by neutralizing metabolically produced “reactive oxygen species” (ROS: H2O −2 , O −2 , OH∙). Left unchecked, hyperoxidation disrupts biomolecules, cellular lipid membranes, organ tissues, metabolic pathways, and genetic mechanisms. Location and function of each tissue type of GPx is specific and not transferable [5]; intracellular glutathione peroxidase-1 (GPx1) resides in most body cells including red blood cells and, as measured in the blood, is a bioindicator of subject Se status.In the Se-adequate host, GPx1 is quiescent and non-essential [5] until chemically attracted to infection-induced ROS [6], for example, inhaled influenza A virus prompts pulmonary cellular production of ROS to attack the virus and to signal initiation of host cellular/biomolecular immune responses to destroy and clear the virus from the body:
-
Influenza virus-laden sneeze → Healthy, Se-adequate host inhales influenza virus → Virus infects lung epithelium → Induces ROS (inflammation) → Chemoattracts GPx1 → Stimulates host proinflammatory chemokines → Induces dendritic cell activity, including polypeptide C1q [7] → Primes viral antigen-specific T cells → ↑ anti-inflammatory interferon-gamma (IFN-γ) and antibody immunoglobulin G (IgG) neutralization of viral antigen → inhibits virus replication and clears virus from host.
Persistence of parasitic viruses requires infecting host cells, pirating host resources, outmaneuvering host immune components and replicating. The generic RNA virus, equipped with a limited genome (3,000–30,000 nucleotides (nt)), codes for antigenic envelope glycoproteins, interior structural and non-structural proteins, and polymerases used for virion replication. Random point mutations, averaging 1–8 × 10−3 nt substitutions per site per year [8, 9], and gene segment re-assortments alter viral modes of operation and, if successful for the virus, effectively outpace human immune system adaptations against the viruses. In the case of influenza A virus, 16 viable hemagglutinin (HA) and seven neuraminidase (NA) glycoproteins have contributed to novel, pandemic influenza A virus types H1N1, H2N2, H3N2, H5N1 [10]. As viral antigenic composition veers from host antibody recognition, the immune system is pressed to combat the particular viral invasion. Thus, commercial vaccines are routinely developed to anticipate and neutralize the various viral antigenic presentations, before pathogenesis.
During Se-deficient host conditions, transcription of GPx1 falls disproportionately to 10% Se-replete levels [11], and the limited nutrient is shunted elsewhere to meet other physiologic functions in Se-requiring tissues [12]. Discrepancy between concurrent heightened immune system requirements for GPx1 during viral infection and its diminished production help define the role of Se in the etiology of VIDs [13]. Initially, host vulnerability to ROS expresses increased bronchial epithelial cell mucus production [14] and lung pathology [15]; T cell proliferation is suppressed and immunocompetence impaired, including poor antibody response [13]. In addition, oxidative stress induces rapid mutation rates in some RNA virus types—often to virulence; under laboratory conditions, these mutations are not stochastic but consistent and quantifiable, and they are immediate, specific, and orders of magnitude faster than for the RNA virus genome under healthy host conditions [16]. Traceability and longevity of these mutations indicates optimal biomolecular reconfiguration for stability and is a basis for viral virulence and pathogenesis.
While researching Keshan disease (KD), a myocarditis endemic to China [17], Beck et al.[18] discovered that women and children infected with Coxsackievirus B3 (CVB3) exhibited this lesioned heart condition when deficient in Se. KD occurs in geologic locales poor in soil Se available to dietary crops, including grains [19], on which inhabitants in rural, isolated regions subsist. Further epidemiology found that benign forms of both CVB3 and influenza virus type A rapidly mutate to virulence in hosts with Se-deficient status [20, 21]. Broome et al. [22] determined that a threshold of 1 μMol Se/L of host blood deterred rapid mutation of a live, attenuated poliomyelitis virus used as vaccine. Though dietary Se supplementation increased host blood Se levels, decreased viral mutation rates for each of these viruses and improved host immune responses including viral clearance from the hosts, mutated virulent RNA virions remained pathogenic and infectious, even to individuals with Se-replete status [20–22].
Method
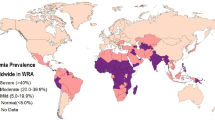
To scope whether the findings of Beck and of Broome [20–22] apply to other VIDs, a world map depicting regions of “low soil-Se” (<0.01 mg/kg) [23] was superimposed with geographic origins of diseases [24] (Fig. 1). Pandemic influenzas type A (H2N2 “Asian,” H3N2 “Hong Kong,” and H5N1 “Avian” influenzas) and severe acute respiratory syndrome (SARS) originated in biogeochemically “Se-poor” regions of China; HIV/AIDS and Ebola hemorrhagic fevers (Ebola “Zaire”, Ebola “Uganda”) originated in nutrient-depleted regions of sub-Saharan Africa (SSA). A literature review of worldwide human blood Se assays indicates sparse data from Se-poor regions of China and SSA, which can fall substantially <1 μMol Se/L (Fig. 2); reported diets there provide as low as 10–17 mcg of Se per day [25, 26], compared to the US recommended daily intake of 55 mcg of Se per day.

Etiological origins of viral infectious diseases correlate with geologic regions of poor Se bioavailability from soils (<0.01 mg/kg, yellow) [23] to food crops. Some countries (U.S.A. and Scandinavia) fertilize with selenium for plant uptake. (Pink patches (China) indicate incidence of Keshan disease; gray ovals depict nutrient iodine deficiency, which seems not to influence the etiology of viral infectious diseases; and brown patches indicate high arsenic concentrations)

To substantiate the above observations, the literature was searched for geographic, ecologic, dietary, cellular, and biomolecular characteristics associated with RNA viruses and VIDs. This review does not consider the protective effects of antioxidant nutrients other than Se, nor any confounding Se antagonists, including toxic concentrations of nutrient iron, methyl-mercury, or other heavy metals, on the etiology of VIDs. Further, this review does not consider the effects of host Se-deficient status on the infectious oncological behavior of tumorigenic viruses such as the human variant of mouse mammary tumor virus or hepatitis virus on hepatoma.
Results
Influenza A Virus
The eight-gene influenza genome encodes 12 known endogenous proteins, including HA/NA surface glycoproteins, three ribonucleic acid (vRNA) polymerase subunits (vRNP: PA, PB1, PB2), non-structural protein (NS1), and matrix proteins M1 and M2. During benign influenza A virus status, the abundant highly conserved matrix M1 protein (M1) transports recently replicated vRNP components from the host cell nucleus to membrane [27, 28]; nonetheless, transport of the viral M1–vRNP assemblage is hindered by host polypeptide C1q [29], housed in infection-responder dendritic cells (DCs). Under healthy host conditions, multifunctional C1q also induces antigen-specific T cell regulation of antibody IgG and cytokine IFN-γ impedance of viral replication [7].
When, however, Nelson et al. [21] infected both Se-adequate and Se-deficient mice with a mild influenza H3N2 virus strain, the influenza A virus matrix M1 gene (M1), not the expected HA or NA glycoprotein genes, mutated rapidly, resulting in a virulent H3N2 variant in the Se-deficient mice; that is, 29 nucleotide (nt) substitutions, within interior nt numbers 309–740 of the M1 segment, code for seven specific aa changes in the newly mutated M1. In this research, Se-deficient-induced ROS is the sole variable resulting in the M1 nucleotide/aa changes. Interior location of the several aa substitutions would alter biomolecular conformation of the M1 structure profoundly, and separate research describes a very different biomolecular mechanism than for the benign M1–C1q interaction [7, 29]. Zhang et al. [30] determined that the N-terminus of M1 firmly binds the globular domain of host C1q, thus blocking C1q stimulus of T cell activity, diminishing T cell use of antibody IgG and induction of INF-γ. As such, virulent M1 subverts DC-C1q functions [30] and the virus evades multiple host immune defenses.
In another mutated influenza A protein example, a single aa substitution (glutamic acid(−) (Glu), lysine(+) (Lys)) at position 627 of the vRNP PB2 subunit correlates with host Se status. Pathogenic avian influenza H5N1 virus [31], with virulent PB2-Lys627 [32], was recovered from dead waterfowl at Lake Qinghai, Qinghai Province, China [33]. While water quality Se assays for Lake Qinghai were not reported, the surrounding geologic landscape is nearly devoid of this essential micronutrient [34, 35]. H5N1-infected dead birds were also found in Hubei Province [36], known for localized regions of poor Se bioavailability [19, 37]; virulence was not detected, however, among asymptomatic H5N1-infected birds of poultry farms and markets of Hong Kong [38]. Asymptomatic bird species infected with influenza A strains 1918 and 2009 H1N1, H2N2, H3N2, and H5N1 carry PB2-Glu627 [27].
Kuzahara et al. [39] determined PB2-Lys+627 lies within an “f-loop” of the PB2 subunit, which they calculate, strengthens PB2 affinity to other vRNP units to increase viral replication. Human pandemic influenza A strains 1918 H1N1, H2N2, H3H2, and H5N1 each contain virulent PB2-Lys627 [27].
Superimposing information of these ROS-induced changes, the immune system cascade becomes interrupted by Se-deficient  host conditions:
host conditions:

SARS Coronavirus
Virulence of SARS in humans begins with pulmonary cell entry of the novel coronavirus, SARS-CoV. The envelope “Spike” (S) glucoprotein of SARS-CoV exhibits two single aa residues, at positions 360 and 479, which both “determine” entry and neutralize host antibody receptor at the host cell surface [40, 41]. Palm civet (Paguma larvata) is the intermediary host for SARS-CoV [42], and civet-CoV retrieved from low-Se Hubei Province more closely relates to human SARS-CoV at determinant aa positions 360 and 479 than does the civet-CoV variant originating from “Se-adequate” Guangdong Province [40]. SARS/civet-like CoV originated in the Chinese horseshoe bat (Rhinolophus macrotis), and genomic sequencing found 71% of the bats assayed in Hubei Province carry the SARS-CoV precursor civet-like coronavirus [43]. That the most successful Spike glucoprotein mutation is retained pre-human infection may exemplify viral “adaptive evolution” stability [44].
HIV
The 1980 origin of pandemic HIV-1/AIDS was traced to specific chimpanzee populations infected with simian immunodeficiency virus (SIVcpz) in southern Cameroon [45]. Contact or use of the infected primates as a food source may have facilitated transfer and evolution of the virus in humans [46].
Asymptomatic, SIV-infected primates carry several viral regulatory genes, including a poorly pathogenic SIV variant of the multifunctional nef gene. Benign nef gene, when transferred via SIVcpz [47] to vulnerable humans, mutates, resulting in virulent nef proteins which interfere with host T cell functions and promote rampant virus replication [48]. Although the host vulnerability condition and genetic/biochemical mechanisms prompting conversion of benign SIV nef variant to HIV-1 virulence are yet to be identified, Hurwitz et al. [49] find that dietary supplementation of 200 mcg Se per day to HIV-infected subjects increased blood Se levels, increased T cell count and decreased viral load. Further, a 5-year randomized, double-blind placebo-controlled clinical trial in Tanzania found a 5% decrease in risk of mortality with each increase of 0.01 μMol Se/L blood from dietary supplementation in HIV-infected pregnant women [50].
EBOV
High mortality (~80%) Ebola hemorrhagic fever erupts episodically (1976–1979, 1994–1996, 2001–2005, 2007), spatially and temporally correlated with droughts [51] and subsequent diminished food production in the Congo Basin [3]. The challenge to find food during lean spells takes human and non-human primates to remote highland fruit bat habitat where several bat species host the Ebola virus (EBOV) [52, 53]. Though outwardly asymptomatic, EBOV-infected bat contact with primates, including humans, is highly virally infectious and contagious, and pathogenic die-off from Ebola hemorrhagic fever during these episodes is severe. The “wild type” (wt) Ebola virus protein 35 (VP35) functions to inhibit host production of interferon transcription factors (INF-3 and INF-7) and aid viral replication [54]; critical to wt VP35 protein virulence are both the interferon inhibitory domain “fold” [54], created by an aa arginine residue at VP35 position 312 [55], and the wt Ebola VP35 use of the host transcription mechanism for interferon production to block immunity against the virus [56].
While the effect of host Se status on Ebola virus virulence is not yet verified, asymptomatic EBOV-infected bats and human survivors of the disease produce virus-specific IgG antibodies [57, 58], whereas T cells and their induced immunoglobulins and INF-γ are undetected in symptomatic EBOV-infected mammals, including humans, which succumb to the systemic disease [59]. This pattern of disabled T cell function is reminiscent of Se-deficiency effects on influenza virus-infected T cells [6, 13–15]. The hunger experienced during the above-described climatic dry spells could be caloric, but if prolonged, also an individual expression of undernutrition and malnutrition, including Se deficiency.
Conclusions
Regions of the globe are void of Se bioavailable from soils for food crop uptake [23] and adequate human nutrition. Worldwide, soils average 0.4 mg Se/kg of soil; “Se-poor” soils in China range between 0.004 and 0.48 mg Se/kg, but Se bioavailability to crops, regardless of soil concentrations, is controlled by biogeochemical factors including soil mineralogy, acidity, oxidation potential, and presence of organic matter [19, 37, 60]. Wide variability in Se accumulation in grains grown on adjacent heterogeneous source rocks in Hubei Province demonstrates this influence [19]:
-
Rice, 0.079, 0.017, 0.063 mg Se/kg grain
-
Wheat, 0.087 0.018, 0.052 mg Se/kg grain
-
Corn, 0.050, 0.015, 0.048 mg Se/kg grain
In Africa, the African Soils Information Service reports degraded, nutrient-depleted conditions for >500 million hectares of SSA soils, and that one third of the population there is “chronically hungry” [61]. For example, though relatively urbanized, and researched, 50% of children in South Africa consume less than half the caloric and nutrient requirements needed for sound health; 70% of a surveyed population “perceive their households to be food insecure and 30% of the households report that their children went to bed hungry, a percentage that increases as incomes decline” [62]. Undernutrition and Se deficiency, especially during pre-natal and childhood periods, have lasting adverse effects which influence physiologic development of immature immune systems and subsequent impaired immunocompetence in post-adolescent years [63, 64]. These dietary and antioxidant Se-deficient nutritional conditions contribute to the physiologic oxidative stress conducive to impaired immune systems and RNA viral mutations, which can be virulent and pathogenic. Table 1 synopsizes the known geographic, ecologic, edaphic, and epidemiologic factors contributing to the VIDs discussed.
The Future
Despite the emergence of the above-described diseases in China and SSA, and the 2007 outbreaks of both poliomyelitis in Nigeria (353 cases) [65] and Ebola “Uganda” [66], etiological origin of infectious RNA viral virulence is not restricted to these geographic regions. Epidemic optic and peripheral neuropathy in Cuba during the early 1990s was traced to infection by otherwise benign Coxsackievirus A9 during an extended period of food shortages, prompting virulence [67].
Etiology of these VIDs suggests that vulnerable populations in any geographic region enduring chronic nutritional Se deprivation, for any reasons, could become spawning grounds for virulence of innate benign but opportunistic RNA viruses. If infectious, the virulent virus can be transferred even to individuals with Se-adequate status. Monitoring incipient hot spots for immune system biomarkers, agricultural productivity, and adequate and nutritious food supply to local populations, is a small order for helping to tame local or global RNA viral virulence.
References
Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3(11):2011–2030
Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K (2003) Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289(2):179–186
Thomson MC, Connor SJ, Ward N, Molyneux D (2004) Impact of climate variability on infectious disease in West Africa. EcoHealth 1:138–150
Beck MA, Handy J, Levander OA (2004) Host nutritional status: the neglected virulence factor. Trends Microbiol 12(9):417–423
Flohe L, Brigelius-Flohe R (2006) In: Hatfield DL, Berry MJ, Gladyshev VN (eds) Selenium: its molecular biology and role in human health. 2nd edition. Springer Science+Business Media, LLC, New York, pp 161–172
Beck MA (2000) Nutritionally induced oxidative stress: effect on viral disease. Am J Clin Nutr 71(suppl):1676S–1679S
Baruah P, Dumitriu IE, Malik TH, Cook HT, Dyson J, Scott D, Simpson E, Botto M (2009) C1q enhances IFN-g production by antigen-specific T cells via the CD40 co-stimulatory pathway on dendritic cells. Blood 113:3485–3493
Nelson MI, Simonsen L, Viboud C, Miller MA, Taylor J, St. George K, Griesemer SB, Ghedi E, Sengamalay NA, Spiro DJ, Volkov I, Grenfell BT, Lipman DJ, Taubenberger JK, Holmes EC (2006) Stochastic processes are key determinants of short-term evolution in influenza A virus. PLoS Pathog 2(12):1144–1151
Holmes EC (2003) Molecular clocks and the puzzle of RNA virus origins. J Virol 77(7):3893–3897
Smith GJD, Bahl J, Vijaykrishna D, Zhang J, Poon LLM, Chen H, Webster RG, Malik Peiris JS, Yi G (2009) Dating the emergence of pandemic influenza viruses. Proc Natl Acad Sci 106(28):11709–11712
Weiss Sachdev S, Sunde RA (2001) Selenium regulation of transcript abundance and translational efficiency of glutathione peroxidase-1 and -4 in rat liver. Biochem J 357:851–858
Bermano G, Nicol F, Dyer JA, Sunde RA, Beckett GJ, Arthur JR, Hesketh JE (1995) Tissue-specific regulation of selenoenzyme gene expression during selenium deficiency in rats. Biochem J 311:425–430
Sheridan PA, Zhong N, Carlson BA, Perella CM, Hatfield DL, Beck MA (2007) Decreased selenoprotein expression alters the immune response during influenza virus infection in mice. J Nutr 137:1466–1471
Jaspers I, Zhang W, Brighton LE, Carson JL, Styblo M, Beck MA (2007) Selenium deficiency alters epithelial cell morphology and responses to influenza. Free Radic Biol Med 42(12):1826–1837
Shrimali RK, Irons RD, Carlson BA, Sano Y, Gladyshev VN, Park JM, Hatfield DL (2008) Selenoproteins mediate T cell immunity through an antioxidant mechanism. J Biol Chem 283(29):20181–20185
Domingo E (1997) Rapid of viral RNA genomes. J Nutr 127:9583–9615
Ge K, Xue A, Bai J, Wang S (1983) Keshan disease—an endemic cardiomyopathy in China. Virchows Arch 401(1):1–15
Beck MA, Esworthy RS, Ho Y-S, Chu F-F (1998) Glutathione peroxidase protects mice from viral-induced myocarditis. FASEB J 12:1143–1146
Wang Z, Gao Y (2001) Biogeochemical cycling of selenium in Chinese environments. Appl Geochem 16:1345–1351
Beck MA, Shi Q, Morris VC, Levander OA (1995) Rapid genomic evolution of a non-virulent Coxsackievirus B3 in selenium-deficient mice results in selection of identical virulent isolates. Nat Med 1:433–436
Nelson HK, Shi Q, van Dael P, Schiffrin EJ, Blum S, Barclay D, Levander OA, Beck MA (2001) Host nutritional selenium status as a driving force for influenza virus mutations. FASEB J 15:1846–1848
Broome CS, McArdle F, Kyle JA, Andrews F, Lowe NM, Hart CA, Arthur JR, Jackson MJ (2004) An increase in selenium intake improves immune function and poliovirus handling in adults with marginal selenium status. Am J Clin Nutr 80:154–164
Oldfield JE (2002) World atlas of selenium. Selenium-Tellurium Association, Grimbergen
Harthill M (2008) Does low Se bioavailability contribute to the emergence of viral infectious diseases? GSA annual meeting, Houston. Abstract. (Iterations >2004)
Alfthan G, Xu GL, Tan WH, Aro A, Wu J, Yang YX, Liang WS, Xue WL, Kong LH (2000) Selenium supplementation of children in a selenium-deficient area in China: blood selenium levels and glutathione peroxidase activities. Biol Trace Elem Res 73(2):113–125
Xia Y, Hill KE, Byrne DW, Xu J, Burk RF (2005) Effectiveness of selenium supplements in a low-selenium area of China. Am J Clin Nutr 81:829–834
Neumann G, Noda T, Kawaoka Y (2009) Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939
Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E et al (50 others) (2009) Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325(5937):197–201
Liu X, Sun L, Yu M, Wang Z, Xu C, Xue O, Zhang K, Ye X, Kitamura Y, Liu W (2009) Cyclophilin A (sic) interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell Microbial 11:730–741
Zhang J, Li G, Liu X, Wang Z, Liu W, Ye X (2009) Influenza A virus M1 blocks the classical complement pathway through interacting with C1qA. J Gen Virol 90:2751–2758
Liu J, Xiao H, Lei F, Zhu Q, Qin K, Zhang X-W, Zhang X-L, Zhao D, Wang G, Feng Y, Ma J, Liu W, Wang J, Gao GF (2005) Highly pathogenic H5N1 influenza virus infection in migratory birds. Science 309:1225–1231
Hatta M, Gao P, Halfmann P, Kawaoka Y (2001) Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293:1840–1842
Wang G, Zhan D, Li L, Lei F, Liu B, Liu D, Xiao H, Feng Y, Li J, Yang B, Yin Z, Song X, Zhu X, Cong Y, Pu J, Wang J, Liu J, Gao GF, Zhu Q (2008) H5N1 avian influenza re-emergence of Lake Qinghai: phylogenetic and antigenic analyses of the newly isolated viruses and roles of migratory birds in virus circulation. J Gen Virol 89:697–702
Song R, Li G, Fen S, Wang Y, Li W, Hiroshi T, Nobum H, Susumu M, Tatsunobu S (2003) Selenium contents in the blood of grazing yaks and rangeland plants in the Eastern Tibet-High Plateau, China. Jap J Livestock Mgmt 39(3):105–1131
Shao S, Zheng B (2008) The biogeochemistry of selenium in Sunan grassland, Gansu, Northwest China, casts doubt on the belief that Marco Polo reported selenosis for the first time in history. Environ Geochem Health 30:307–314
Yu Z, Song Y, Zhou H, Xu X, Hu Q, Wu H, Zhang A, Zhou Y, Chen J, Dan H, Luo Q, Li X, Chen H, Jin M (2007) Avian influenza (H5N1) virus in waterfowl and chickens, Central China. Emerg Infect Dis 13(5):772–775
Fordyce FM, Zhang G, Green K, Liu X (2000) Soil, grain and water chemistry in relation to human selenium-responsive diseases in Enshi District, China. Appl Geochem 15:117–132
Smith GJD, Vijaykrishna D, Ellis TM, Dyrting KC, Leung YHC, Bahl J, Wong CW, Kai H, Chow MKW, Duan L, Chan ASL, Zhang LJ, Chen H, Luk GSM, Peiris JSM, Guan Y (2009) Characterization of avian influenza viruses A (H5N1) from wild birds, Hong Kong, 2004–2008. Emerg Infect Dis 15(3):402–407
Kuzuhara T, Kise D, Yoshida H, Horita T, Murazaki Y, Nishimura A, Echigo N, Utsunomiya H, Tsuge H (2009) Structural basis of the influenza A virus RNA polymerase PB2 RNA-binding domain containing the pathogenicity-determinant lysine 627 residue. J Biol Chem 284(11):6855–6860
Liu L, Fang Q, Deng F, Wang H, Yi CE, Ba L, Yu W, Lin RD, Li T, Hu Z, Ho DD, Zhang L, Chen Z (2007) Natural mutations in the receptor binding domain of spike glycoprotein determine the reactivity of cross-neutralization between palm civet coronavirus and severe acute respiratory syndrome coronavirus. J Virol 81(9):4694–4700
Yi CE, Ba L, Zhang L, Ho DD, Chen Z (2005) Single amino acid substitutions in the severe acute respiratory syndrome coronavirus spike glycoprotein determine viral entry and immunogenicity of a major neutralizing domain. J Virol 79(18):11638–11646
Song HD, Tu CC, Zhang GW, Wang SY, Zheng K, Lei LC, Chen QX, Gao YW, Zhou HQ, Xiang H, Zheng HJ, Chern SW, Cheng F, Pan CM, Xuan H, Chen SJ, Luo HM, Zhou DH, Liu YF, He JF, Qin PZ, Li LH, Ren YQ, Liang WJ, Yu YD, Anderson L, Wang M, Xu RH, Wu XW, Zheng HY, Chen JD, Liang G, Gao Y, Liao M, Fang L, Jiang LY, Li H, Chen F, Di B, He LJ, Lin JY, Tong S, Kong X, Du L, Hao P, Tang H, Bernini A, Yu XJ, Spiga O, Guo ZM, Pan HY, He WZ, Manuguerra JC, Fontanet A, Danchin A, Niccolai N, Li YX, Wu CI, Zhao GP (2005) Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and humans. Proc Natl Acad Sci USA 102:2430–2435
Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang G, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF (2005) Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679
Zhang C-Y, Wei J-F, He S-H (2006) Adaptive evolution of the spike gene of SARS coronavirus: changes in positively selected sites in different epidemic groups. BMC Microbiol 6:88–97
Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JFY, Sharp PM, Shaw GM, Peeters M, Hahn BH (2006) Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313:523–526
LeBreton M, Yang O, Tamoufe U, Mpoudi-Ngole E, Torimio JN, Djoko CF, Carr JK, Prosser AT, Rimoin AW, Birx DL, Burke DS, Wolfe ND (2007) Exposure to wild primates among HIV-infected persons. Emerg Infect Dis 13(10):1579–1582
Homann S, Tibroni N, Baumann I, Sertel S, Keppler OT, Fackler OT (2009) Determinants in HIV-1 Nef for enhancement of virus replication and depletion of CD4+ T lymphocytes in human lymphoid tissue ex vivo. Retrovirology 6(6), 14 pages http://www.retrovirology.com/content/6/1/6)
Arhel N, Lehmann M, Clauβ K, Nienhaus GU, Piguet VJ, Kirchhoff F (2009) The inability to disrupt the immunological synapse between infected human T cells and APCs distinguishes HIV-1 from most other primate lentiviruses. J Clin Invest 119(10):2965–2975
Hurwitz BE, Klaus JR, Llabre MM, Gonzalez A, Lawrence PJ, Maher KJ, Greeson JM, Baum MK, Shor-Posner G, Skyler JS, Schneiderman N (2007) Suppression of human immunodeficiency virus type 1 viral load with selenium supplementation. Arch Intern Med 167:148–154
Kupka R, Msamanga GI, Spiegelman D, Morris S, Mugusi F, Hunter DJ, Fawzi WW (2004) Selenium status is associated with accelerated HIV disease progression among HIV-1-infected pregnant women in Tanzania. J Nutr 134:2556–2560
Leroy EM, Rouquet P, Formenty P, Souquiere S, Kilbourne A, Froment J-M, Bermejo M, Smit S, Karesh W, Swanepoel R, Zaki SR, Rollin PE (2004) Multiple Ebola virus transmission events and rapid decline of Central African wildlife. Science 303:387–392
Biek R, Walsh PD, Leroy EM, Real LA (2006) Recent common ancestry of Ebola Zaire virus found in a bat reservoir. PLoS Pathog 2(10):885–886
Wittmann TJ, Biek R, Hassanin A, Rouquet P, Reed P, Yaba P, Pourrut X, Real LA, Gonzalez J-P, Leroy EM (2007) Isolates of Zaire ebolavirus from wild apes reveal genetic lineage and recombinants. Proc Natl Acad Sci 104(43):17123–17127
Leung DW, Ginder ND, Fulton DB, Nix J, Basler CF, Honzatko RB, Amarasinghe GK (2009) Structure of the Ebola VP35 interferon inhibitory domain. Proc Natl Acad Sci 106(2):411–416
Hartman AL, Bird BH, Towner JS, Antoniadou Z-A, Zak SR, Nichol ST (2008) Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of Ebola virus. J Virol 82(6):2699–2704
Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, Ozato K (2009) Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog 5(6):e1000493, 15 pages
Pourrut X, Souris M, Towner JS, Rollin PE, Nichol ST, Gonzalez JP, Leroy EM (2009) Large serological survey showing cocirculation of Ebola and Marburg viruses in Gabonese bat populations, and a high seroprevalence of both viruses in Rousettus aegyptiacus. BMC Infect Dis 9:159–168
Wauquier N, Becquart P, Glasquet C, Leroy EM (2009) Immunoglobulin G in Ebola outbreak survivors, Gabon. Emerg Infect Dis 15(7):1136–1137
Warfield KL, Olinger G, Deal EM, Swenson DL, Bailey M, Negley DL, Hart MK, Bavari S (2005) Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection. J Immun 175:1184–1191
Johnson CC, Ge X, Green KA, Liu X (2000) Selenium distribution in the local environment of selected villages of the Keshan Disease belt, Zhangjiakou District, Hebei Province, Peoples’ Republic of China. Appl Geochem 15(3):385–401
African Soils Information Service (AfSIS). Summary of AfSIS Launch Media Coverage, Nairobi, Kenya, January 13, 2009. http://www.africasoils.net
Hunter N, May J, Padayachee V (2003) Lessons for PRSP from Poverty Reduction Strategies in South Africa, 3rd Meeting of the African Meeting Group on the Poverty Reduction Strategy Papers, Addis Ababa, Ethiopia, Economic Commission for Africa, 40 pages
McDade TW, Beck MA, Kuzawa C, Adair LS (2001) Prenatal undernutrition, postnatal environments, and antibody response to vaccination in adolescence. Am J Clin Nutr 74:543–548
Cunningham-Rundles S, McNeely DF, Moon A (2005) Mechanisms of nutrient modulation of the immune response. J Allergy Clin Imunol 115(6):1235–1237
World Health Organization (2009) World health statistics, Table 3 Selected infectious diseases, pp 59–69
Towner JS, Sealy TK, Khristova ML, Albarino CG, Conlan S, Reeder P-LQ, Lipkin WI, Downing R, Tappero JW, Okware S, Lutwama J, Bakamutumaho B, Kayiwa J, Comer JA, Rollin PE, Kasiazek TG, Nichol ST (2008) Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog 4(11):1–6
Beck MA, Matthews CC (2000) Micronutrients and host resistance to viral infection. Proc Nutr Soc 59:581–585
Reid AH, Fanning TG, Janczewski TA, McCall S, Taubenberger JK (2002) Characterization of the 1918 “Spanish” influenza virus matrix gene segment. J Virology 76(21):10717–10723
Yu H, Zhang GH, Hua RH, Zhang Q, Lui TQ, Liao M, Tong GZ (2007) Isolation and genetic analysis of human origin H1N1 and H3N2 influenza viruses from pigs in China. Biochem Biophys Res Commun 356(1):91–96
Zeng X, Hua Y, Li X, Zhang Z (2008) Monitoring influenza A virus and Newcastle disease virus in migratory waterfowls in Sanjiang natural reserve of Heilongjiang Province. Wei Sheng Wu Xue Bao 48(10):1403–1407
Santiago ML, Range F, Keele BF, Li Y, Bailes E, Bibollet-Ruche F, Fruteau C, Noe R, Peeters M, Brookfield JFY, Shaw GM, Sharp PM, Hahn BH (2005) Simian immunodeficiency virus infection in free-ranging sooty mangabeys (Cercocebus atys atys) from the Taï Forest, Côte d’Ivoire: implications for the origin of epidemic human immunodeficiency virus Type 2. J Virology 79(19):12515–12527
Levander OA (1986) In: Mertz W (ed) Trace elements in human and animal nutrition. Academic, London, pp 139–197
Arnaud J, Malvy D, Richard M-J, Faure H, Chaventré A (2001) Selenium status in an iodine deficient population of the West Ivory Coast. J Physiol Anthropol 20(2):81–84
Fondu P, Hariga-Muller C, Mozes N, Neve J, Van Steirteghem A, Mandelbaum IM (1978) Protein-energy malnutrition and anemia in Kivu (Zaire). Am J Clin Nutr 30:46–56
Ngo DB, Dikassa L, Okitolonda W, Kashala TD, Gervy C, Dumont J, Vanovervelt N, Contempré B, Diplock AT, Peach S, Vanderpas J (1997) Selenium status in pregnant women of a rural population (Zaire) in relationship to iodine deficiency. Trop Med Intl Health 2(6):572–581
Benemariya H, Robberecht H, Deelstra H (1993) Daily dietary intake of copper, zinc and selenium by different population groups in Burundi, Africa. Sci Total Environ 136(1–2):49–76
Diplock AT (1993) Indices of selenium status in human populations. Am J Clin Nutr 57(suppl):256S–258S
Van Lettow M, Harries AD, Kumwenda JJ, Zijlstra EE, Clark TD, Taha TE, Semba RD (2004) Micronutrient malnutrition and wasting in adults with pulmonary tuberculosis with and without HIV co-infection in Malawi. BMC Infect Dis 4:61–69
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Harthill, M. Review: Micronutrient Selenium Deficiency Influences Evolution of Some Viral Infectious Diseases. Biol Trace Elem Res 143, 1325–1336 (2011). https://doi.org/10.1007/s12011-011-8977-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-011-8977-1