
Abstract
Background
Bone marrow-derived mesenchymal stem cells (bmMSCs) have been used as a cellular therapeutic option for treatment of osteonecrosis of the femoral head. However, use of bmMSCs as a treatment adjuvant for orthopaedic disorders in general has achieved limited success. Adipose-derived MSCs (aMSCs) may be a more-efficient regenerative cell source given their greater quantity and protection from physiologic stress.
Questions/purposes
We asked the following questions in a paired analysis of MSCs from patients with osteonecrosis: (1) Is there a difference in proliferation potential between aMSCs and bmMSCs? (2) Is there a difference in osteogenic differentiation potential between aMSCs and bmMSCs? (3) Are genetic pathways differentially expressed between aMSCs and bmMSCs that may govern functional phenotypic discrepancies?
Methods
Periarticular samples of adipose tissue and bone marrow from the femoral canal were obtained from 15 patients undergoing hip replacement for late-stage (Steinberg Stages III-VI) osteonecrosis. MSCs were isolated from both tissue sources and taken through a standardized 20-day cell division protocol to establish cumulative cell count. They also were grown in osteogenic differentiation media for 14 days with subsequent measurement of alkaline phosphatase in units of optical density. RNA was isolated from aMSCs and bmMSCs in five patients to assess differentially expressed genetic pathways using the Affymetrix GeneChip® Human Transcriptome Array 2.0 platform.
Results
Proliferation capacity was increased by fourfold in aMSCs compared with bmMSCs after 20 days in culture. The mean difference in cumulative cell count was 3.99 × 108 cells (SD = 1.67 × 108 cells; 95% CI, 3.07 × 108–4.92 × 108 cells; p < 0.001). Bone differentiation efficiency as measured by optical density was increased by 2.25-fold in aMSCs compared with bmMSCs. The mean difference in optical density was 1.27 (SD = 0.34; 95% CI, 1.08–1.46; p < 0.001). RNA transcriptome analysis showed 284 genes that met statistical (p < 0.05) and biological (fold change > 1.5) significance cutoffs for differential expression between cell populations. Subsequent network topology of differentially expressed genes showed alterations in pathways critical for musculoskeletal tissue development in addition to many nonspecific findings.
Conclusions
aMSCs outperform bmMSCs in growth rate and bone differentiation potential in the setting of osteonecrosis, suggesting they may provide a more-potent regenerative therapeutic strategy in this population. Differential expression of genes and cellular pathways highlighted in this study may provide therapeutic targets for cellular optimization or acellular treatment strategies.
Clinical Relevance
aMSCs may provide a more robust cellular therapeutic than bmMSCs for treatment of osteonecrosis. Ideally, a well-designed prospective study will be able to evaluate the efficacy of these cellular therapies side-by-side in patients with bilateral early stage disease.
Similar content being viewed by others

Introduction
Osteonecrosis of the femoral head is a disease that may be amenable to regenerative therapeutic strategies if intervention occurs during early stages [13]. Mesenchymal stem cells (MSCs) are promising cellular candidates for regenerative orthopaedics because of their ability to differentiate to several tissues of mesodermal origin including bone [11, 38]. To date, bone marrow-derived mesenchymal stem cells (bmMSCs) have been used as a regenerative therapeutic for treatment of osteonecrosis, osteoarthritis, bony defects, and coating of implants [3, 8, 15, 19, 21–23, 25, 29, 30]. However, treatment of orthopaedic disorders with bmMSCs has achieved limited success [19, 21, 22]. This likely is partly because bone marrow is a tissue subjected to high levels of physiologic stress and cell turnover [6]. Furthermore, bmMSCs have been shown to lose potency with age and certain disease states like osteoporosis [10, 26]. Finally, the concentration of bmMSCs in the bone marrow is modest compared with other sources in the body [6].
Adipose-derived MSCs (aMSCs) are thought to be a potentially more-efficient regenerative cell source given their greater quantity and protection from physiologic stress relative to bmMSCs [2, 6, 28, 38]. Furthermore, these cells have a low immunogenicity profile and are capable of immunomodulation, increasing their clinical applicability [16, 24]. aMSCs currently are being investigated in several clinical trials for multiple diseases including osteoarthritis (NCT01739504; NCT01585857), cartilage defects (NCT02090140), craniofacial bone and soft tissue injury (NCT01633892), Crohn’s disease (NCT01157650; NCT01011244), urinary incontinence (NCT01799694; NCT01804153), graft versus host disease (NCT01222039), and multiple sclerosis (NCT0143764) (http://clinicaltrials.gov/) [5]. Although aMSCs certainly are more abundant than bmMSCs and hold theoretical promise for regenerative medicine, further phenotypic characterization is required to determine applicability in specific disease states. Therefore, we sought to compare regenerative performance metrics between aMSCs and bmMSCs in the setting of osteonecrosis and explore potential mechanisms for observed differences.
We asked the following specific questions in a paired analysis of MSCs from patients with osteonecrosis of the femoral head: (1) Is there a difference in proliferation potential between aMSCs and bmMSCs? (2) Is there a difference in osteogenic differentiation potential between aMSCs and bmMSCs? (3) Are genetic pathways differentially expressed between aMSCs and bmMSCs that may govern functional phenotypic discrepancies?
Patients and Methods
Human Tissue Collection and Processing
After institutional review board approval, 2 g periarticular adipose tissue and 5 cc femoral canal bone marrow were obtained from 15 patients undergoing primary THA for late-stage (Steinberg Stages III-VI) osteonecrosis (Table 1). Adipose tissue was processed as previously described [37]. Briefly, adipose tissue was minced with a surgical scalpel and incubated in 0.01% collagenase Type I (Sigma Aldrich, St Louis, MO, USA) for 90 minutes. The digested adipose tissue then was centrifuged, washed, and strained to separate tissue debris; then incubated in red blood cell lysis buffer (Stem Cell Technologies, Vancouver, BC, Canada). The derived solution was centrifuged, supernatant removed, and the pellet resuspended in expansion media containing advanced minimum essential medium (aMEM) with 10% fetal bovine serum, 100 µmol/L penicillin, 100 g/mol/L streptomycin, and 2 mol/L-glutamine (Invitrogen, Carlsbad, CA, USA). Femoral canal bone marrow was washed with phosphate-buffered saline (Corning Inc, Manassas, VA, USA) and centrifuged at 1700 revolutions per minute for 5 minutes. The resultant pellet was then treated with red cell lysis buffer for 10 minutes at 0 °C, resuspended and cultured in expansion media.
MSC Culture
Cells were grown on 100-mm culture dishes at 37 °C in a humidified 5% CO2 incubator. Twenty-four hours after the initial plating, cultures were gently washed with phosphate-buffered saline to remove any nonadherent cells and replaced with expansion media. Every 48 hours the culture medium was replaced completely and nonadherent cells were discarded. MSCs were identified as having the ability to proliferate in culture, with adherent, spindle-shaped morphologic features. Cells were expanded until they reached 90% confluence, at which time they were trypsinized and expanded until the second passage.
Immunophenotypic Characterization of Cells
After the second passage in cell culture, a subset of cells was attained for immunophenotypic analysis. Trypsinized cells were washed in medium and centrifuged for 5 minutes at 1700 revolutions per minute to create a cell pellet. The cell pellet then was resuspended in 100 μL phosphate-buffered saline and removed for immunophenotypic analysis. Each cell preparation was stained with the following monoclonal antibodies: mouse antihuman CD34-APC (clone MOPC-21), CD14-FITC (clone G155-178), CD45-PE-Cy7 (clone MOPC-21) (all from BD Biosciences, San Jose, CA, USA), CD105-PE (clone SN6), and CD90-PerCP-Cy5.5 (clone 5E10) (both from eBioscience, San Diego, CA, USA). Analysis was performed using an LSR II flow cytometer, and the subsequent data were analyzed with Cell Quest Pro software (both from BD Biosciences).
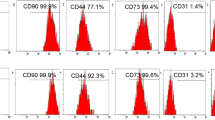
Immunophenotypic analysis of aMSCs and bmMSCs showed a high proportion of cells that were CD 90 and CD 105 positive, while simultaneously being CD 14, CD 34, and CD 45 negative; this combination of cell markers is consistent with MSC phenotype (Fig. 1).

Mesenchymal stem cells were isolated from periarticular adipose tissue and femoral canal bone marrow. After the third passage, cells in culture were analyzed with phenotypic immunostaining via fluorescent activated cell sorting. (A) aMSCs are CD 90 and CD 105 positive. (B) aMSCs are CD 45 and CD 14 negative. (C) aMSCs are CD 45 and CD 34 negative. (D) bmMSCs are CD 90 and CD 105 positive. (E) bmMSCs are CD 45 and CD 14 negative. (F) bmMSCs are CD 45 and CD 34 negative. Collectively, these results show that a high proportion of CD 105/90 positive and CD 14/34/45 negative cells were isolated from adipose tissue and iliac crest bone marrow, which is consistent with MSC phenotype.
Histologic Characterization of Cells
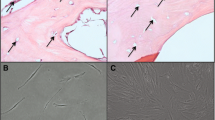
After the second passage in culture a subset of cells was obtained to confirm, in a qualitative fashion, that aMSCs and bmMSCs were capable of trilineage differentiation to bone, cartilage, and adipose tissue. Osteogenic, chondrogenic and adipogenic differentiation were achieved using StemPro® Osteogenesis, Chondrogenesis, and Adipogenesis differentiation media, respectively (Invitrogen), with 10% fetal bovine serum per the manufacturer protocol. Cells were cultured for a total of 8 weeks with media changed every 48 hours. After 8 weeks, cells grown in osteogenic medium were stained with 2% alizarin red (Sigma-Aldrich), cells grown in chondrogenic medium were stained with 1% Alcian blue (Sigma-Aldrich), and cells grown in adipogenesis medium were stained with 0.2% Oil Red-O (Sigma-Aldrich). Cells were observed using a light microscope (Carl Zeiss Microscopy, Thornwood, NY, USA) with resultant images acquired at ×20 magnification (Fig. 2).

aMSCs and bmMSCs were capable of trilineage musculoskeletal differentiation. After the third passage, cells in culture were grown in either osteogenic, chondrogenic, or adipogenic media for 8 weeks and subsequently stained with alizarin red, Alcian blue, or Oil Red-O, respectively. Microscopy was performed at ×20 magnification. (A) Osteogenic, (B) chondrogenic, and (C) adipogenic differentiation are shown from aMSCs. (D) Osteogenic, (E) chondrogenic, and (F) adipogenic differentiation are shown from bmMSCs.
Assessment of Cell Proliferation
After the second passage in culture, aMSCs and bmMSCs were subjected to a standardized cell proliferation protocol as follows. On Day 0, MSCs were quantified using a CountessTM (Invitrogen) automated cell counter. The CountessTM detects viable and nonviable cells using 0.4% trypan blue (Invitrogen) staining. Three-hundred thousand cells were plated on 100-mm culture dishes with expansion medium. On the second day, the expansion medium was changed and on Day 4 cells were trypsinized and subsequently counted. From the counted cells, 300,000 were isolated and replated on new 100-mm dishes. This procedure was replicated four times for a total protocol of 20 days from which cell proliferation metrics could be quantified. Cumulative population doubling was calculated using the formula: [log10 (NH) − log10(N1)]/log102, where NH is the harvested cell number and N1 is the plated cell number [4]. Each passage was calculated and added to the population doubling of the previous passage to obtain the cumulative population doubling. The generation time (ie, the time between two cell doublings) was calculated as: [Log102 × Δt]/[log10(NH) − log10 (N1)], where Δt is the time between passages [4]. Cumulative cell count was the number of cells generated during the 20-day proliferation protocol.
Assessment of Osteogenic Differentiation
Osteogenic differentiation was achieved using StemPro® Osteogenesis differentiation medium (Invitrogen) with 10% fetal bovine serum, per the manufacturer protocol. Ten-thousand MSCs were plated in a 12-well culture dish. Cells were cultured for a total of 14 days with the medium changed every 48 hours. After Day 14, osteogenic differentiation was measured through proxy quantification of alkaline phosphatase. Alkaline phosphatase activity was measured using an Alkaline Phosphatase Assay Kit (Abcam, Cambridge, MA, USA) per the manufacturer protocol. This colorimetric assay was reported in units of optical density.
Transcriptome Profiling
Transcriptome mRNA discrepancy in MSC lineages was assessed. RNA was extracted from five patients’ aMSCs and bmMSCs after the third passage in culture (RNAeasy Kit; Qiagen, Valencia, CA, USA). Affymetrix 3’ IVT Express Kit (Affymetrix, Santa Clara, CA, USA) was used as the microarray platform performed as per manufacturer recommendation. One-hundred nanograms of high-quality total RNA (Agilent Bioanalyzer; Agilent Technologies Inc, Santa Clara, CA, USA) was used for reverse transcription with products column-purified (Affymetrix) and transcribed to yield biotin-labeled cRNA hybridized onto Affymetrix U133 Plus 2.0 GeneChips®. Arrays washed and stained with streptavidin-phycoerythrin were scanned in an Affymetrix GeneChip® Scanner 3000. Control parameters confirmed standard ranges before normalization, noise correction, and data reduction. Transcriptome analysis was performed (GeneSpring GX 12.1; Agilent Technologies) with probes filtered according to preset flag values, yielding a volcano plot to identify significant gene expression based on pairwise comparison at fold change and Benjamini-Hochberg false discovery rate corrected p value (> 1.5-fold, p < 0.05).
Network Analysis
Molecular interactions of differentially expressed genes between aMSCs and bmMSCs were examined to identify pathways of relevance in the initial list of root objects (Ingenuity® Pathways Analysis and the MetaCore database; Qiagen) [6]. Overlap between imported datasets and canonical pathways imparted significance as determined by hypergeometric distribution analysis. Internetwork prioritization based on imported datasets was measured by z-score = (r − μ)/σ, ranking subnetworks according to saturation with root objects. Finally, confidence interval evaluation of network relevance to gene ontology biologic processes was measured as p value = ((R!n!(N − R)!(N − n)!)/N!)Σ [min(n,R), i = max(r,R + n − N)]((1)/(i!(R − i)!(n − i)!(N − R − n + i)!)) [6].
Statistical Analysis
Results were expressed as means or differences in means with 95% CI. Analysis was performed through paired Student’s t-tests using JMP® 9 software (SAS, Cary, NC, USA). All statistical tests were two-sided and the threshold for statistical significance was set at α = 0.05.
Results
Assessment of Proliferation Potential
Proliferation capacity was increased in patient-matched aMSCs compared with bmMSCs. Cumulative cell count was higher in aMSCs at every time and fourfold greater at 20 days; the mean difference in cumulative cell count at 20 days was 3.99 × 108 cells (SD = 1.67 × 108 cells; 95% CI, 3.07 × 108–4.9 2 × 108 cells; p < 0.001). Generation time in days was shorter in aMSCs; the mean difference in generation time was 0.43 days (SD = 0.16 days; 95% CI, 0.34–0.52 days; p < 0.001). Cumulative population doubling was greater in aMSCs compared with bmMSCs at passage one (2.42 versus 1.74; p < 0.001), passage two (6.65 versus 4.97; p < 0.001), passage three (8.98 versus 6.39; p < 0.001), passage four (10.98 versus 8.87; p < 0.001), and passage five (13.10 versus 10.35; p < 0.001) (Fig. 3).

aMSCs show superior proliferation potential compared with bmMSCs. After the third passage, cells in culture were plated at a concentration of 300,000 per 10-cm dish. They were grown for 96 hours after which time they were counted and replated at a concentration of 300,000 per 10-cm dish. This process was repeated five times to derive growth curves. (A) Cumulative population doubling was increased in aMSCs compared with bmMSCs at each passage (p < 0.001). (B) The cumulative cell count was fourfold greater in aMSCs compared with bmMSCs at the end of the 20-day protocol (p < 0.001). (C) Generation time was less in aMSCs compared with bmMSCs (p < 0.001). pts = patients.
Assessment of Osteogenic Differentiation Potential
Bone differentiation efficiency as measured by optical density was increased in patient-matched aMSCs compared with bmMSCs. Optical density was 2.25-fold higher in aMSCs at 14 days; the mean difference in optical density was 1.27 (SD = 0.34; 95% CI, 1.08–1.46; p < 0.001). Optical density was greater in aMSCs for all 15 patients in the study (Fig. 4).

aMSCs show increased osteogenic differentiation potential compared with bmMSCs. After the third passage, cells in culture were grown in osteogenic differentiation media for 14 days. Alkaline phosphatase activity was measured as optical density between aMSCs and bmMSCs from the same 15 patients (pts). (A) Optical density was increased in aMSCs for all 15 patients. (B) aMSCs showed a 2.5-fold increase in optical density across the entire cohort (p < 0.001).
Genetic Transcriptome Analysis
Gene expression differed between patient-matched MSCs depending on native tissue source. RNA transcriptome analysis showed 284 genes that met statistical (p < 0.05; false discovery rate corrected) and biological (fold change > 1.5) significance cutoffs for differential expression between aMSCs and bmMSCs. The top 50 most differentially expressed genes all showed fold changes of 5.5 or greater (Table 2). Subsequent network analysis of these 284 genes determined the most interconnected biological pathways (Fig. 5A). The top 10 molecular pathways implicated in the network of differentially expressed genes included YWHAZ, SUMO1, ERBB2, CDK2, NR3C1, H3, AR, CAND1, SP1, HSP90 (Fig. 5B). These genes have been associated with processes such as response to glucocorticoids, response to androgens, response to insulin, calcium homeostasis, cell growth, metabolism, immunity, and inflammation (Table 3). Further network topology analysis of differentially expressed genes showed alterations in processes critical for musculoskeletal tissue development and many nonspecific pathways (Fig. 5C).

Differentially expressed genetic pathways govern functional phenotypic differences between aMSCs and bmMSCs. (A) This network diagram shows the relationships among differentially expressed genes between aMSCs and bmMSCs. The expression of 54,000 genes was measured by the Affymetrix GeneChip® Human Transcriptome Array 2.0 platform with 284 genes meeting cutoffs for biologic significance (fold change > 1.5) and statistical significance (p < 0.05). Subsequent network interactions were performed with Ingenuity® Pathway Analysis software. (B) Network topology was performed on all 284 differentially expressed genes and their biologic pathway relationships. The number of connections to a molecular pathway is represented on the x-axis as  (degrees). The top 10 most implicated pathways are shown in the pink square. (C) This graph shows the top 10 ontologic processes governed by differentially expressed genes. Notably, skeletal and muscular system development and function, connective tissue development and function, and hematologic system development and function achieved high levels of significance.
(degrees). The top 10 most implicated pathways are shown in the pink square. (C) This graph shows the top 10 ontologic processes governed by differentially expressed genes. Notably, skeletal and muscular system development and function, connective tissue development and function, and hematologic system development and function achieved high levels of significance.
Discussion
Orthopaedic surgery is entering a new era where biologic treatment modalities are beginning to complement and even replace mechanical solutions of the past. MSCs have been a cornerstone of this effort for diseases such as osteonecrosis of the femoral head with autologous bmMSCs being the most commonly used cellular therapeutic approach. However, to date regenerative medicine with bmMSCs has not proven to be a panacea for patients [9, 21, 22]. Studies are beginning to suggest that aMSCs may provide a more-robust source of stem cells given their relative protection from physiologic stress [2, 6, 28, 38]. These cells also are more abundant and accessible than bmMSCs. Nevertheless, questions remain regarding the osteogenic potential of aMSCs, particularly as it pertains to specific disease states. Therefore, the goals of our study were to characterize the proliferation and osteogenic differentiation potential between aMSCs and bmMSCs from patients with osteonecrosis, while exploring if differential genetic expression exists based on mesenchymal stem cell lineage.
The results of this study must be considered in light of important limitations. First, this was an in vitro laboratory investigation. As such, it is difficult to fully recapitulate the in vivo microenvironment that will modulate activity of these cells. The efficacy of these cellular therapeutic strategies will need to be assessed in prospective human studies before firm conclusions can be drawn. However, the phenotypic differences from this investigation in conjunction with the success of aMSCs for other therapeutic indications are promising indicators. Second, without experimental manipulation of the differentially expressed genes identified by transcriptome analysis, we cannot comment on how specific genes potentially modify phenotypic performance. Nevertheless, the purpose of this exploratory analysis was to determine if genes are differentially expressed that may underlie functional differences, and identify candidates for future study in the laboratory. Third, our transcriptome analysis did not identify differential expression of some primary regulators of osteogenesis such as osteocalcin, runt-related transcription factor 2, and BMP-2. Transcriptome profiling was done on predifferentiated MSCs. Identified differentially expressed genes likely regulate the expression of these important osteogenic pathways; however, further transcriptome profiling on predifferentiated and osteogenically differentiated MSCs will be needed to elucidate more precise relationships. Fourth, network biology analytic techniques are restricted by gene functions reported to date. Therefore, our study contributes to the growing repertoire of gene ontology data and will strengthen the platform for future investigations of musculoskeletal genetics, development, and physiology that leverage network techniques.
Hip decompression and implantation of bmMSCs has been reported to be a successful treatment for early stage osteonecrosis. There is a subgroup of patients however, for whom surgical treatment has failed [22]. Reported predictors of failure include size of the necrotic lesion, etiology of the osteonecrosis, and proliferation potential of the bmMSCs based on fibroblast colony-forming units [13, 22]. In addition, previous studies have shown that patients with corticosteroid-induced and alcohol-induced osteonecrosis have decreased osteogenesis from their bmMSCs [7, 9, 12, 17, 34] and could account for the limited results obtained in certain osteonecrosis groups. Identifying other potential sources of MSCs is of critical importance for management of patients who may be considered at high risk for failure.
Our study showed increased proliferation capacity among aMSCs in comparison to bmMSCs from patients with osteonecrosis. This finding is consistent with those of numerous studies corroborating this phenomenon in in vitro and animal studies [1, 5, 6, 8, 33]. Strioga et al. [33] reported that this may be attributable to fewer in vivo cell division events for aMSCs, resulting in longer telomere lengths and greater reserve capacity. aMSCs seem to be buffered from physiologic insult as residents of a quiescent tissue, whereas bmMSCs are subject to a dynamic environment constantly adapting to homeostatic derangements. Our study shows that in the setting of osteonecrosis, aMSCs maintain an enhanced capacity for growth relative to MSCs isolated from bone marrow. These data should lead to future prospective studies exploring the clinical differences between aMSCs and bmMSCs in differing groups of patients with osteonecrosis.
Despite having a proliferation advantage, numerous studies have shown inferior osteogenesis from aMSCs compared with bmMSCs [14, 18, 20, 27, 31, 35]. These studies from in vitro experiments and animal models have questioned the ability of aMSCs for regenerative orthopaedics. Nevertheless, other work has shown equal or superior osteogenesis from aMSCs when compared with bmMSCs [32, 36, 38]. Although this debate remains unclear it will be critical to evaluate candidate stem cells from the specific patient population to be treated. For example, bmMSCs have been shown to lose potency with age and certain disease states like osteoporosis [10, 26]. Likewise, our results showed that in the setting of osteonecrosis, aMSCs showed superior osteogenic potential compared with bmMSCs as measured by alkaline phosphatase activity. This may be explained partly in previous studies showing that patients with corticosteroid-induced and alcohol-induced osteonecrosis have decreased osteogenesis from their bmMSCs [7, 9, 12, 17, 34]. Greater physiologic stress in the bone marrow may contribute to the discrepant osteogenesis profile of MSCs based on native tissue source in patients with these insults. These differences are especially important for surgeons treating osteonecrosis, and may have an even greater effect in patients still in contact with the etiologic risk factor that led to its occurrence (ie, continued steroid therapy), further encouraging the surgeon to choose aMSCs over bmMCSs at the time of hip decompression.
Only one published study, to our knowledge, has delineated alterations in the transcriptome of aMSCs and bmMSCs [1]. However, Abu Kasim et al. [1] compared the transcriptome of aMSCs, bmMSCs, and MSCs from Wharton’s Jelly and dental pulp. They found limited expression profile differences, but did find that aMSCs had more prominent expression of genes involved in tissue development, whereas bmMSCs favored inflammation and immunomodulation pathways. These findings are consistent with those of our study, which also highlighted few gene expression differences, but noted a propensity toward tissue development pathways in aMSCs (Fig. 5). What seems apparent is that MSCs from different tissues exhibit distinctive gene expression signatures making them competent for lineage-specific differentiation. As such, MSC source should be an important consideration based on patient characteristics, disease state, and desired final differentiated tissue type. The results of our study support phenotypic advantage for aMSCs in osteonecrosis of the femoral head, which may be explained by a combination of factors including physiologic protection from insults and a baseline genetic expression profile with a proclivity toward tissue development. The most interconnected pathways from the network analysis in our study included the glucocorticoid receptor, androgen receptor, and other proteins regulating insulin sensitivity, cell growth, metabolism, and protein function. Corticosteroid use is a primary risk factor for osteonecrosis; therefore, alterations in the glucocorticoid receptor between aMSCs and bmMSCs may partly explain osteoregeneration discrepancies in patients with osteonecrosis. Followup investigation will be necessary to determine the extent to which these identified genes and pathways govern osteonecrosis pathophysiology and if these pathways are viable therapeutic targets.
We found that aMSCs maintain greater potential for proliferation and osteogenic differentiation compared with bmMSCs in the setting of osteonecrosis. Patients with early-stage disease are increasingly receiving joint-preserving operations such as core decompression in combination with autologous bone marrow toward the goal of preventing collapse of the femoral head and regenerating the hip. As aMSCs are more abundant and show a superior functional phenotype for this purpose, they may prove to be a more effective therapeutic approach. We also identified differentially expressed genes between aMSCs and bmMSCs, potentially providing the basis for further exploration in disease pathophysiology and potential therapeutic targets. Studies that selectively modify these identified genes and subsequently test cellular performance metrics will be critical in this endeavor. Nevertheless, as more centers gain the infrastructure to use aMSCs, patients with osteonecrosis may realize greater benefit from this autologous cellular therapeutic strategy. Ideally, a well-designed prospective study will be able to evaluate the efficacy of these cellular therapies side-by-side in patients with bilateral early-stage disease.
References
Abu Kasim NH, Govindasamy V, Gnanasegaran N, Musa S, Pradeep PJ, Srijaya TC, Aziz ZA. Unique molecular signatures influencing the biological function and fate of post-natal stem cells isolated from different sources. J Tissue Eng Regen Med. 2012 Dec 10. [Epub ahead of print].
Abudusaimi A, Aihemaitijiang Y, Wang YH, Cui L, Maimaitiming S, Abulikemu M. Adipose-derived stem cells enhance bone regeneration in vascular necrosis of the femoral head in the rabbit. J Int Med Res. 2011;39:1852–1860.
Aoyama T, Goto K, Kakinoki R, Ikeguchi R, Ueda M, Kasai Y, Maekawa T, Tada H, Teramukai S, Nakamura T, Toguchida J. An exploratory clinical trial for idiopathic osteonecrosis of femoral head by cultured autologous multipotent mesenchymal stromal cells augmented with vascularized bone grafts. Tissue Eng Part B Rev. 2014;20:233–242.
Bieback K, Hecker A, Kocaomer A, Lannert H, Schallmoser K, Strunk D, Kluter H. Human alternatives to fetal bovine serum for the expansion of mesenchymal stromal cells from bone marrow. Stem Cells. 2009;27:2331–2341.
Casteilla L, Planat-Benard V, Laharrague P, Cousin B. Adipose-derived stromal cells: their identity and uses in clinical trials, an update. World J Stem Cells. 2011;3:25–33.
Crespo-Diaz R, Behfar A, Butler GW, Padley DJ, Sarr MG, Bartunek J, Dietz AB, Terzic A. Platelet lysate consisting of a natural repair proteome supports human mesenchymal stem cell proliferation and chromosomal stability. Cell Transplant. 2011;20:797–811.
Cui Q, Wang GJ, Balian G. Steroid-induced adipogenesis in a pluripotential cell line from bone marrow. J Bone Joint Surg Am. 1997;79:1054–1063.
Deans RJ, Moseley AB. Mesenchymal stem cells: biology and potential clinical uses. Exp Hematol. 2000;28:875–884.
Gangji V, De Maertelaer V, Hauzeur JP. Autologous bone marrow cell implantation in the treatment of non-traumatic osteonecrosis of the femoral head: five year follow-up of a prospective controlled study. Bone. 2011;49:1005–1009.
Giannoudis P, Tzioupis C, Almalki T, Buckley R. Fracture healing in osteoporotic fractures: is it really different? A basic science perspective. Injury. 2007;38(suppl 1):S90–99.
Gimble J, Guilak F. Adipose-derived adult stem cells: isolation, characterization, and differentiation potential. Cytotherapy. 2003;5:362–369.
Hernigou P, Beaujean F. Abnormalities in the bone marrow of the iliac crest in patients who have osteonecrosis secondary to corticosteroid therapy or alcohol abuse. J Bone Joint Surg Am. 1997;79:1047–1053.
Houdek MT, Wyles CC, Martin JR, Sierra RJ. Stem cell treatment for avascular necrosis of the femoral head: current perspectives. Stem Cells Cloning. 2014;7:65–70.
Im GI, Shin YW, Lee KB. Do adipose tissue-derived mesenchymal stem cells have the same osteogenic and chondrogenic potential as bone marrow-derived cells? Osteoarthritis Cartilage. 2005;13:845–853.
Jin H, Xia B, Yu N, He B, Shen Y, Xiao L, Tong P. The effects of autologous bone marrow mesenchymal stem cell arterial perfusion on vascular repair and angiogenesis in osteonecrosis of the femoral head in dogs. Int Orthop. 2012;36:2589–2596.
Kronsteiner B, Wolbank S, Peterbauer A, Hackl C, Redl H, van Griensven M, Gabriel C. Human mesenchymal stem cells from adipose tissue and amnion influence T-cells depending on stimulation method and presence of other immune cells. Stem Cells Dev. 2011;20:2115–2126.
Lee JS, Lee JS, Roh HL, Kim CH, Jung JS, Suh KT. Alterations in the differentiation ability of mesenchymal stem cells in patients with nontraumatic osteonecrosis of the femoral head: comparative analysis according to the risk factor. J Orthop Res. 2006;24:604–609.
Liao HT, Chen CT. Osteogenic potential: comparison between bone marrow and adipose-derived mesenchymal stem cells. World J Stem Cells. 2014;6:288–295.
Lim YW, Kim YS, Lee JW, Kwon SY. Stem cell implantation for osteonecrosis of the femoral head. Exp Mol Med. 2013;45:e61.
Liu TM, Martina M, Hutmacher DW, Hui JH, Lee EH, Lim B. Identification of common pathways mediating differentiation of bone marrow- and adipose tissue-derived human mesenchymal stem cells into three mesenchymal lineages. Stem Cells. 2007;25:750–760.
Mao Q, Jin H, Liao F, Xiao L, Chen D, Tong P. The efficacy of targeted intraarterial delivery of concentrated autologous bone marrow containing mononuclear cells in the treatment of osteonecrosis of the femoral head: a five year follow-up study. Bone. 2013;57:509–516.
Martin JR, Houdek MT, Sierra RJ. Use of concentrated bone marrow aspirate and platelet rich plasma during minimally invasive decompression of the femoral head in the treatment of osteonecrosis. Croat Med J. 2013;54:219–224.
Matsuya H, Kushida T, Asada T, Umeda M, Wada T, Iida H. Regenerative effects of transplanting autologous mesenchymal stem cells on corticosteroid-induced osteonecrosis in rabbits. Mod Rheumatol. 2008;18:132–139.
McIntosh K, Zvonic S, Garrett S, Mitchell JB, Floyd ZE, Hammill L, Kloster A, Di Halvorsen Y, Ting JP, Storms RW, Goh B, Kilroy G, Wu X, Gimble JM. The immunogenicity of human adipose-derived cells: temporal changes in vitro. Stem Cells. 2006;24:1246–1253.
Minguell JJ, Conget P, Erices A. Biology and clinical utilization of mesenchymal progenitor cells. Braz J Med Biol Res. 2000;33:881–887.
Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell. 2004;3:379–389.
Niemeyer P, Fechner K, Milz S, Richter W, Suedkamp NP, Mehlhorn AT, Pearce S, Kasten P. Comparison of mesenchymal stem cells from bone marrow and adipose tissue for bone regeneration in a critical size defect of the sheep tibia and the influence of platelet-rich plasma. Biomaterials. 2010;31:3572–3579.
Pak J. Regeneration of human bones in hip osteonecrosis and human cartilage in knee osteoarthritis with autologous adipose-tissue-derived stem cells: a case series. J Med Case Rep. 2011;5:296.
Rastogi S, Sankineani SR, Nag HL, Mohanty S, Shivanand G, Marimuthu K, Kumar R, Rijal L. Intralesional autologous mesenchymal stem cells in management of osteonecrosis of femur: a preliminary study. Musculoskelet Surg. 2013;97:223–228.
Schwartz RE, Reyes M, Koodie L, Jiang Y, Blackstad M, Lund T, Lenvik T, Johnson S, Hu WS, Verfaillie CM. Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest. 2002;109:1291–1302.
Shafiee A, Seyedjafari E, Soleimani M, Ahmadbeigi N, Dinarvand P, Ghaemi N. A comparison between osteogenic differentiation of human unrestricted somatic stem cells and mesenchymal stem cells from bone marrow and adipose tissue. Biotechnol Lett. 2011;33:1257–1264.
Stockmann P, Park J, von Wilmowsky C, Nkenke E, Felszeghy E, Dehner JF, Schmitt C, Tudor C, Schlegel KA. Guided bone regeneration in pig calvarial bone defects using autologous mesenchymal stem/progenitor cells: a comparison of different tissue sources. J Craniomaxillofac Surg. 2012;40:310–320.
Strioga M, Viswanathan S, Darinskas A, Slaby O, Michalek J. Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev. 2012;21:2724–2752.
Suh KT, Kim SW, Roh HL, Youn MS, Jung JS. Decreased osteogenic differentiation of mesenchymal stem cells in alcohol-induced osteonecrosis. Clin Orthop Relat Res. 2005;431:220–225.
Vishnubalaji R, Al-Nbaheen M, Kadalmani B, Aldahmash A, Ramesh T. Comparative investigation of the differentiation capability of bone-marrow- and adipose-derived mesenchymal stem cells by qualitative and quantitative analysis. Cell Tissue Res. 2012;347:419–427.
Wen Y, Jiang B, Cui J, Li G, Yu M, Wang F, Zhang G, Nan X, Yue W, Xu X, Pei X. Superior osteogenic capacity of different mesenchymal stem cells for bone tissue engineering. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:e324–332.
Wyles CC, Houdek MT, Wyles SP, Wagner ER, Behfar A, Sierra RJ. Differential cytotoxicity of corticosteroids on human mesenchymal stem cells. Clin Orthop Relat Res. 2015;473:1155–1164.
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13:4279–4295.
Acknowledgments
We thank Lois A. Rowe BS (Mayo Clinic Center for Regenerative Medicine), for assistance with histologic preparation and analysis and D. Kent Arrell PhD (Mayo Clinic Center for Regenerative Medicine), for assistance with transcriptome analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported by an Accelerated Regenerative Medicine Grant from the Mayo Clinic Center for Regenerative Medicine (Rochester, MN, USA). This publication was made possible by CTSA Grant Number UL1 TR000135 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
The institution of one or more of the authors (RJS) has received, during the study period, funding from the Mayo Clinic Center for Regenerative Medicine (Rochester, MN, USA). One of the authors certifies that he (RJS), or a member of his immediate family, has received or may receive payments or benefits, during the study period, an amount of USD 10,000 to USD 100,000 from Biomet Inc (Warsaw, IN, USA).
All ICMJE Conflict of Interest Forms for authors and Clinical Orthopaedics and Related Research ® editors and board members are on file with the publication and can be viewed on request.
Each author certifies that his or her institution approved or waived approval for the human protocol for this investigation and that all investigations were conducted in conformity with ethical principles of research.
About this article

Cite this article
Wyles, C.C., Houdek, M.T., Crespo-Diaz, R.J. et al. Adipose-derived Mesenchymal Stem Cells Are Phenotypically Superior for Regeneration in the Setting of Osteonecrosis of the Femoral Head. Clin Orthop Relat Res 473, 3080–3090 (2015). https://doi.org/10.1007/s11999-015-4385-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11999-015-4385-8