
Opinion statement
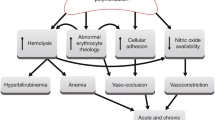
Current treatment options for stroke in sickle cell disease (SCD) and thalassemia are limited. Hypercoagulation occurs in both diseases partly due to activated platelets and red blood cell dysmorphology and dysfunction, resulting in chronic anemia. This overlapping pathophysiology of the nervous system promotes the role of some common treatment modalities for these similar diseases. The current evidence suggests that chronic exchange transfusion and stem cell transplantation/bone marrow transplant (BMT) can be used in both diseases. Exchange transfusion is the mainstay of therapy of acute stroke in SCD whereas blood transfusions and hydroxyurea appear to be the most effective current treatments. However, evidence suggests that exchange transfusion should be initiated in acute ischemic stroke (AIS) and chronic transfusion continued in both diseases after AIS. Exchange transfusion can also be used acutely in AIS with thalassemia as this disorder is also associated with hypervolemia at baseline, occurring secondary to chronic anemia. The ideal length of chronic transfusions for both primary and secondary stroke prevention still needs to be better defined. Stem cell transplant or BMT is the only curative treatment for both diseases. However, timing needs to be further investigated. If transplantation is effective, it may need to be done before the child with SCD expresses disease, such as in infancy. However, in infancy, we cannot predict the severity of the phenotype in SCD with certainty, so an individual decision about transplantation is difficult to make. In thalassemia, transplantation may be effective later because vasculopathy is not the problem as in SCD. Furthermore, cerebrovascular disease occurs later in thalassemia than in SCD. Finally, aspirin is a treatment modality that also warrants further investigation. There are limited studies on the effectiveness of aspirin in SCD and thalassemias. Few studies have demonstrated clinical improvement of stroke in patients with hemoglobinopathies. Given the successful use of aspirin in the treatment and prevention of recurrent cardioembolic events in patients without hemoglobinopathies, diseases with hypercoagulability, such as SCD and thalassemia, may also benefit from the use of aspirin for treatment and prevention. However, the evidence available is based on case and retrospective studies, necessitating future larger and more valid studies to evaluate safety and effectiveness.
Similar content being viewed by others


References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Prengler M, Pavlakis S, Prohovnik I, Adams R. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51:543–52.
Prohovnik I, Hurlet-Jensen A, Adams R, et al. Hemodynamic etiology of elevated flow velocity and stroke in sickle-cell disease. J Cereb Blood F Met. 2009;29:803–10.
Yuan J, Bunyaratvej A, Fucharoen S, et al. The instability of the membrane skeleton in thalassemic red blood cells. Blood. 1995;86(10):3945.
Succar J, Khalid M, Taher AT. Perspectives: Thalassemia and venous thromboembolism. Mediterr J Hematol Infect Dis. 2011; 3.
Galanello R, Origa R. Review: Beta-thalassemia. Orphanet J Rare Dis. 2010; 5(11).
Taher A, Isma’eel H, Mehio G, et al. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost. 2006;96(4):488–91.
Belisario AR, Rodrigues CV, Martins ML, et al. Coinheritance of α-thalassemia decreases the risk of cerebrovascular disease in a cohort of children with sickle cell anemia. Hemoglobin. 2010;34(6):516–29.
Vineeta S, Biswas A, Kumar B, Saxena R. Protein C and Protein S: causative factor for developing a hemorrhagic infarct in a HbE/Beta thalassemia child. Indian J Pediatr. 2010;77(3):316–7.
Bishop S, Matheus MG, Abboud MR, et al. Effect of chronic transfusion therapy on progression of neurovascular pathology in pediatric patients with sickle cell anemia. Blood Cells Mol Dis. 2011;47(2):125–8.
Vermeer SE, Prins ND, den Heijer T, et al. Silent brain infarcts and the risk of dementia and cognitive decline. N Eng J Med. 2003;348:1215–22.
Vassilopoulou S, Anagnostou E, Paraskevas G, Spengos K. Etiology and treatment of ischaemic stroke in patients with β-thalassemia major. Eur J Neurol. 2011;18:1426–8.
Ware RE, Schultz WH, Yovetich N, et al. Stroke with transfusions changing to Hydroxyurea (SWiTCH): a phase III randomized clinical trial for treatment of children with sickle cell Anemia, Stroke, and iron overload. Pediatr Blood Cancer. 2011;57(6):1011–7.
Ware RE: Transcranial Dopplers (TCD) with Transfusions Changing to Hydroxyurea. Accessed online at ClinicalTrials.gov on 3 Nov 2011. This is somewhat a follow-up from the SWITCH trial, with the same investigators, and equivalent patient population. The main difference here is that the primary outcome is TCD abnormalities, and the secondary outcome is strokes. The trial is currently in progress.
Lee MT, Piomelli S, Granger S, et al. Stroke prevention trial in sickle cell Anemia (STOP): extended follow-up and final results. Blood. 2006;108(3):847–52.
Musallam KM, Taher AT. Thrombosis in Thalassemia: why are we so concerned? Hemoglobin. 2011;35(5–6):503–10.
Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity. Blood. 2010;115(10):1886–92.
Taher AT, Mussalam KM, Karimi M, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost. 2010;8:2152–8.
Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle-cell anemia and abnormal results on transcranial Doppler ultrasound. N Eng J Med. 1998;339(1):5–11.
Sarode R, Matevosyan K, Rogers ZR, et al. Advantages of isovolemic hemodilution – red cell exchange therapy to prevent recurrent stroke in sickle cell anemia patients. J Clin Apheresis. 2011;26(4):200–7.
Lucarelli G, Gaziev J, Isgro A, et al. Allogenic Cellular Gene Therapy in Hemoglobinopathies – Evaluation of Hematopoietic SCT in Sickle Cell Anemia. Bone Marrow Transpl. 2011; E-published ahead of print 18 April 2011. This is one of the most recently published articles on stem cell transplants in SCD patients. It is well known and accepted that transplantation in these patients is curative, as is shown in this study, with excellent long-term follow-up. It remains to be examined which patients are ideal candidates and the optimal time for these procedures to be effective.
Oberoi S, Bansal D, Singh P, Marwaha RK. Stroke in a young boy with β-thalassemia intermedia secondary to Moyamoya syndrome. J Pediatr Hematol Oncol. 2010;32:568–70.
Dhouib N, Mellouli F, Ouederni M, et al. Early onset of cerebrovascular accident in a thalassemia major child. Tunis Med. 2010;88(5):372.
Furie KL, Kasner SE, Adams RJ, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227–76.
Disclosure
No conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ali, N., Srey, R. & Pavlakis, S. Hemoglobinopathies and Stroke: Strategies for Prevention and Treatment. Curr Treat Options Cardio Med 14, 227–236 (2012). https://doi.org/10.1007/s11936-012-0173-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11936-012-0173-x